


BIOVIA Materials Studio是一个完整的建模和模拟环境,旨在让材料科学和化学研究人员预测和理解材料的原子和分子结构与其特性和行为之间的关系。使用材料工作室,许多行业的研究人员正在设计性能更好的各类材料,包括催化剂、聚合物、复合材料、金属、合金、制药、电池等。

 最新更新
最新更新
1. CASTEP对自旋极化(磁性)体系可以进行振动特性的线性响应计算;
2. CASTEP的DFT+U不再需要对非磁性系统进行自旋极化设置,使其计算速度至少快了两倍;
3. 在隐式溶剂存在的情况下,可以使用CASTEP进行力计算。进而可以在执行考虑溶剂影响的任务中进行几何优化、分子动力学计算。
1. DFTB+服务器已更新到版本 22.1;
2. FlexTS 中的 NEB 路径阈值现在可以确保复杂反应途径的稳定收敛;
3. 新的CERIA和TiO2纳米参数设置为DFTB+中一些其他的金属氧化物提供了支持;
4. COSMO 隐式溶剂化模型现已在 DFTB+ 中可用;
5. 该模块中的FlexTS 路径轨迹文件现在可以按照正确的顺序提供。
1. 该模块中的FlexTS 路径轨迹文件现在可以按照正确的顺序提供;
2. FlexTS 中的 NEB 路径阈值现在可以确保复杂反应途径的稳定收敛。
1. Forcite GPU 计算现在支持嵌入式原子模型;
2. 改进了COMPASSIII水模型计算扩散系数预测方法;
3. Forcite分析现在支持从密度场计算结构因子;
4. Forcite分析现在支持各向异性分量的压强分析;
5. 对Forcite 非键和动力学 GPU 算法的更新令性能显著增强。
1. GULP 服务器已更新到版本 6.1;
2. 一个新的universal forcefield GFN-FF的周期性版本现在可以在GULP中使用。如J. Gale等人在J. Chem.Theory Comput. 17 (2021) 7827–7849中描述该力场是一个具有接近量子力学精度的力场;
3. 现在可以在GULP中拟合到八阶的双体相互作用的多项式势项。
1. 现在可以在NVIDIA GPUs上运行Mesocite耗散粒子动力学(DPD)计算;
2. Mesocite分析现在支持各向异性分量的压力分析;
3. Mesocite分析现在支持从密度场计算结构因子;
4. Mesocite 的DPD现在支持使用高斯分布计算的珠子电荷静电势。
1. ONETEP 服务器已升级到版本 6.1.12;
2. ONETEP中的杂化交换相关函数实现扩展到涵盖三维周期系统,而不仅仅是“盒子中的分子”几何化。
产品特点
1、加快创新进程:加深对定义材料属性的相互作用的理解。BIOVIA Materials Studio使材料科学家和研究团队能够比单独进行测试和实验更快,更高效地开发新的,性能更好,成本更低的材料。
2、降低研发成本:通过"虚拟筛选"候选人尽量减少物理实验的数量。Materials Studio的客户已经表示,引入新材料所需的实验数量减少了10倍。
3、提高研发效率:在管道试点中实现自动化并共享更佳实践,以减少非增值任务。通过创建可重复使用的建模和仿真协议来自动执行重复性或单调乏味的建模任务。
4、促进数据驱动决策:用强大的材料信息学补充实验室实验
5、协作:捕捉和分享专业知识和方法,使计算科学在组织和地理边界上更加一致。
6、解决您最困难的问题:BIOVIA专家科学家的工作人员确保及时的支持和专业知识,帮助解决材料科学中较具挑战性的问题。
系统与硬件要求
| 项目 | 推荐配置 | 浮动许可证服务器系统 |
|---|---|---|
| 操作系统 | Windows 10 | Windows sever |
| CPU | Intel Core i5或 更高 | Intel Core i5或 更高 |
| 内存 | 8G+ RAM | 8GB+RAM |
| 存储 | 512GB 7200转 SATA硬盘 | 512GB 7200转 SATA硬盘 |
| 显卡 | Nvidia or AMD 512MB显卡 | Nvidia or AMD 512MB显卡 |
| 显示器 | 1920*1080分辨率显示器 | 1920*1080分辨率显示器 |
| 其他 |
- |
- |
产品说明
|
Materials visualizer |
模块配置 | |||
|
BIOVIA Materials Studio 材料模拟软件以可视化视窗界面为核心 |
CASTEP | DMol3 | DFTB+ | ONETEP |
| AmorphousCell | ForcitePlus | COMPASS | GULP | |
| Mesocite | Sorption | Synthia | QSAR | |
| Polymorph | Morphology | Reflex | 共计22个模块 | |
探索材料属性
材料工作室提供"以硅为先"的方法,使研究人员能够在物理测试之前,在成本相对较低的环境中优化材料的性能。
- 加快创新进程:加深对定义材料属性的相互作用的理解
- 降低研发成本:通过"虚拟筛选"候选人尽量减少物理实验的数量
- 提高研发效率:在管道试点中实现自动化并共享更佳实践,以减少非增值任务
- 促进数据驱动决策:用强大的材料信息学补充实验室实验

BIOVIA Materials Studio,许多行业的研究人员正在设计性能更好的各种材料,包括制药,催化剂,聚合物和复合材料,金属和合金,电池和燃料电池等等。
多尺度和多物理解决方案
探索下面的功能领域,了解研究人员如何开发更好的催化剂、药物、聚合物、复合材料、合金、电池等。
使用Materials Studio,您可以:
加速创新:BIOVIA Materials Studio使材料科学家和研究团队能够比单独进行测试和实验更快,更高效地开发新的,性能更好,成本更低的材料。
降低成本:Materials Studio的客户已经表示,引入新材料所需的实验数量减少了10倍。
提高效率:通过创建可重复使用的建模和仿真协议来自动执行重复性或单调乏味的建模任务。
协作:捕捉和分享专业知识和方法,使计算科学在组织和地理边界上更加一致。
解决您最困难的问题:BIOVIA专家科学家的工作人员确保及时的支持和专业知识,帮助解决材料科学中较具挑战性的问题。
Materials Studio包含一个图形用户环境 - Materials Studio Visualizer,研究人员可以在其中构建,操作和查看分子模型,结晶材料,表面,聚合物和中尺度结构。材料工作室Visualizer补充了一整套解决方法,包括量子,原子(或“古典”),中尺度和统计,使研究人员能够评估不同粒径和时间尺度的材料。它还包括评估晶体结构和晶体生长的工具。
Materials Studio与BIOVIA Pipeline Pilot集成
Materials Studio与BIOVIA的开放,可扩展和科学意识的平台集成在一起,使其成为业界在材料建模和仿真领域较具吸引力的价值。专家科学家可以使用BIOVIA Pipeline Pilot及其科学收藏(包括Materials Studio Collection)封装和自动化可重用“协议”中的更佳实践。它也可以与第三方或内部应用程序集成,以扩展Materials Studio支持的科学的广度和深度。
Materials Studio内置量子力学,分子力学,介观模拟,机器学习,统计分析和结晶学等门类齐全的仿真功能。它拥有丰富多样的不同尺度仿真能力,让研究人员能够在各种粒子大小尺度和时间尺度预测材料性能,有效平衡计算时间与计算效率。
量子力学工具
Materials Studio CANTERA
Cantera 是一种化学反应速率方程求解器。Materials Studio Cantera为化学反应速率的求解提供热力学输入和运行环境。通过Cantera Reaction Editor,用户能够创建新的反应物种和反应类型,并基于Materials Studio DMol3 获取反应相关信息,并将反应类型、物种、反应速率等信息加入已有反应机理文件中。
Materials Studio CASTEP
Materials Studio CASTEP基于平面波-赝势密度泛函方法,能够模拟包括陶瓷、半导体和金属材料在内的多种材料的固体、界面和表面性质。
Materials Studio DMol3
Materials Studio DMol3基于原子轨道线性组合密度泛函方法,能够模拟有机/无机分子、团簇、分子晶体、共价键晶体、金属晶体和无限表面的电子结构和材料性质。
Materials Studio DFTB+
Materials Studio DFTB+基于紧束缚的半经验密度泛函方法,融合了密度泛函方法准确性和紧束缚方法高效性,能够在较大系统规模上实现量子力学精度的模拟。
Materials Studio NMR CASTEP
Materials Studio NMR CASTEP依据第一性原理模拟NMR化学位移和电场梯度张量。能够进行分子核磁和固体核磁的计算,适用于陶瓷和半导体等多种材料体系。
Materials Studio ONETEP
Materials Studio ONETEP是一款线性标度的密度泛函程序,能在对多达数千个原子的体系进行第一性原理计算。
Materials Studio QMERA
Materials Studio QMERA采用QM/MM量子力学与分子力学杂化的方法,兼具量子力学计算的准确性与经典力学计算的高效率。这种方法能够处理超大体系的关键位点精确计算,大幅度节省计算量。
Materials Studio VAMP
Materials Studio VAMP使用半经验分子轨道方法,能够快速预测分子有机/无机系统的众多物理/化学特性。Materials Studio VAMP是一种理想的居于经典力场方法和第一性原理方法之间的折中。
经典模拟工具
Materials Studio提供极为丰富的基于原子和分子间经典相互作用的模拟工具,包括分子动力学、晶格动力学和多种蒙特卡洛方法,并提供多种可选择力场。
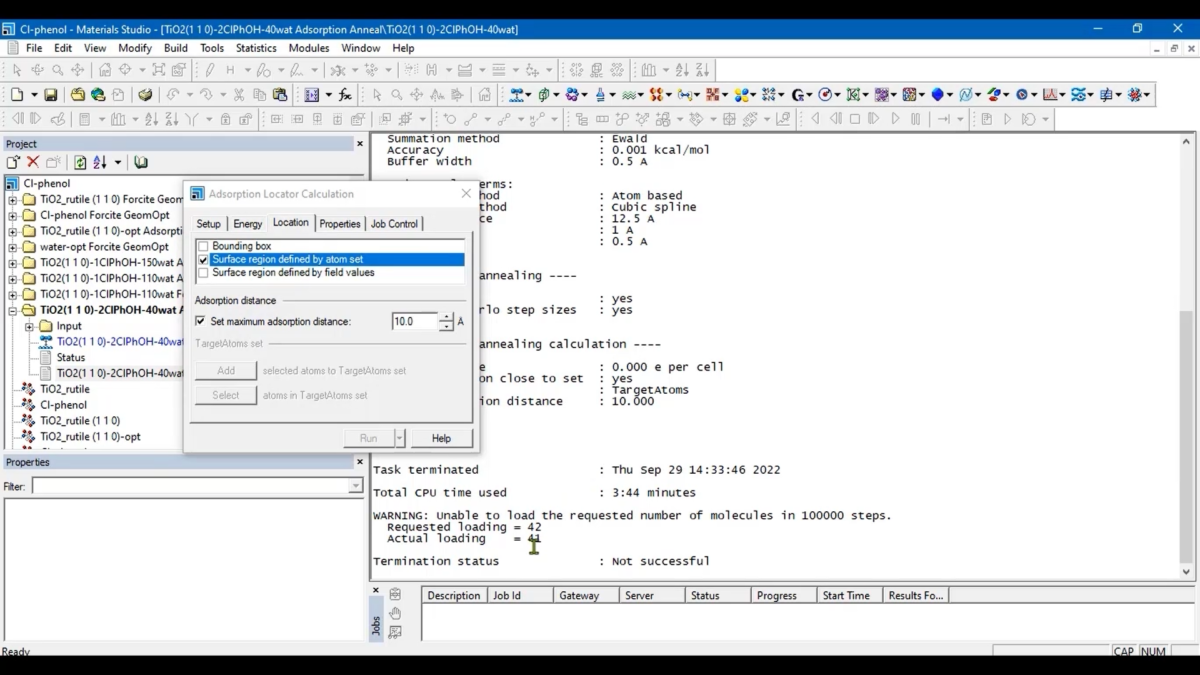
Materials Studio Adsorption Locator
Materials Studio Adsorption Locator是一款采用蒙特卡罗模拟退火方法搜索吸附质在基底材料上的最低能量吸附构象的工具。
Materials Studio Amorphous Cell
Materials Studio Amorphous Cell是一款采用蒙特卡洛方法搭建复杂无定形模型的工具。
Materials Studio Blends
Materials Studio Blends采用扩展的Flory-Huggins模型估算二元混合物体系相容性,能够处理溶剂-溶剂、聚合物-溶剂以及聚合物-聚合物等体系。
Materials Studio Conformers
Materials Studio Conformers提供构象搜索算法和分析工具,用于表征化分子构象和柔性。
Materials Studio COMPASS
Materials Studio COMPASS是一个功能强大的第一性原理力场,能够精确预测单分子和凝聚态体系的结构,构象,震动和热力学性质,并适用于广大的温度和压力区间。
Materials Studio Forcite Plus
Forcite Plus为周期性和非周期性体系提供分子力学和动力学方法。该工具具备丰富的分析功能,能够预测力学性能、扩散率、局部结构、密度变化、内聚能密度、偶极自相关函数等材料属性。支持力场包括Materials Studio COMPASS、CVFF、PCFF、Dreiding和Universal等。
Materials Studio GULP
GULP是一款用于材料优化、属性计算和动力学模拟的工具。它包含众多针对金属、氧化物、矿物,半导体和共价键系统的专用力场。并提供了专有的力场拟合工具,方便进行力场开发。
Materials Studio Sorption
Sorption为研究吸收和分离现象提供基本属性预测方法,能够模拟吸附等温线和亨利常数。
介观模拟工具
Materials Studio内的介观模拟基于粗粒化模型,将经典模型中的若干原子视为一个基本结构单元,等效为一个珠子。这类方法能在比经典模拟更大的体系和更长的时间尺度上进行模拟仿真。
MesoDyn
MesoDyn基于动态平均场密度泛函方法,能够在大尺度和长周期范围内研究尤其适用于复杂聚合物系统的相位分离和结构。
Materials Studio Mesocite
Mesocite基于粗粒化模型,能够处理长度尺度从纳米到微米,时间尺度从纳秒到微秒的材料体系。Materials Studio Mesocite能够提供流体在剪切,限制剪切和平衡条件下的结构和动力学特性。
统计学工具
统计学工具将分子特性直接与实验观测量联系起来,是快速筛选化合物的理想方法。
Materials Studio QSAR
Materials Studio内集成的构效定量关系(QSAR)功能可调用大量描述符和高级分析功能,有助于生成高质量的结构-活性关系,它通过构建材料的实验信息(“性质”)和分子水平特征(“描述符”)之间的统计回归模型,进而预测未知材料的性质。QSAR包含有拓扑描述符和电子结构-拓扑描述符在内的多种描述符。并且,内置的Jurs描述符能够评价溶剂表面的电荷分布;内置的VAMP描述符进一步扩大3D描述符的范围,使之能表达电子相互作用。还支持GFA算法,采用先进的基因算法能计算结构-活性间的定量关系。
Materials Studio QSAR Plus
QSAR Plus增添DMol3描述符,能够为QSAR计算反应性指数和基于量子力学的高精度能量指标。此外,还提供神经网络算法,可用于构建非线性模型,并能够提高模型对噪声数据的耐受度,也可用于有缺值的数据集,和用于构建预测多种物理属性的加权模型。
Materials Studio Synthia
Synthia使用先进的构效定量关系(QSPR)方法计算均聚物和共聚物的属性。能够快速筛选具有某些特性的聚合物种类或共聚物配比。
谱图分析和结晶工具
谱图分析和结晶工具用于研究、预测和修订晶体结构和晶体生长。
Materials Studio Morphology
Morphology基于晶体的原子结构预测晶体形貌。Morphology能够预测晶体形状,分析晶面稳定性,开发定制添加剂以及控制溶剂和杂质对晶体形貌的影响。
Materials Studio QSAR Plus
QSAR Plus增添DMol3描述符,能够为QSAR计算反应性指数和基于量子力学的高精度能量指标。此外,还提供神经网络算法,可用于构建非线性模型,并能够提高模型对噪声数据的耐受度,也可用于有缺值的数据集,和用于构建预测多种物理属性的加权模型。
Materials Studio Polymorph Predictor
Polymorph Predictor是一个以力场为基础,采用蒙特卡洛模拟退火法,由给定化合物的分子结构预测其多晶型的工具。能够用于主要由碳、氮、氧、氢构成的有足够刚性的非离子分子或离子分子,在选定空间点群内基于晶格能量极小来搜索可能的分子堆积排列。
Materials Studio Motif
Motif分析分子晶体中的连接信息,为氢键拓扑提供定性定量分析方法。与Polymorph的预测功能结合,Motif能够为预测结构进行结构分类和打分。与剑桥晶体学数据中心的CSD剑桥结构数据库对接后,它能调用Mercury功能。
Materials Studio Reflex
Reflex用于根据晶体材料模型仿真X射线、中子和电子粉末衍射谱图。Reflex Plus为根据中高质量粉末衍射数据判定晶体结构提供了一整套完整软件包。
Materials Studio Reflex QPA
Reflex QPA扩展了Reflex的功能,能够进行物相分析,可根据粉末衍射数据,确定混合物中各个单相(包括无机系统和有机系统)的相对比例。
Materials Studio X-Cell
X-Cell是一种用于中高质量粉末衍射数据的高效率索引算法。X-Cell使用优化的两分法流程穷举搜索参数空间,为所有可能的单位晶格解建立完整列表。
-
请问如何用径向分布函数(RDF)分析耗散分子力学(DPD)模拟胶束自组装中胶束质心的RDF呢
-
 2023-11-16 11:51:21
2023-11-16 11:51:21欢迎联系我们或拨打24H热线:400-886-8338
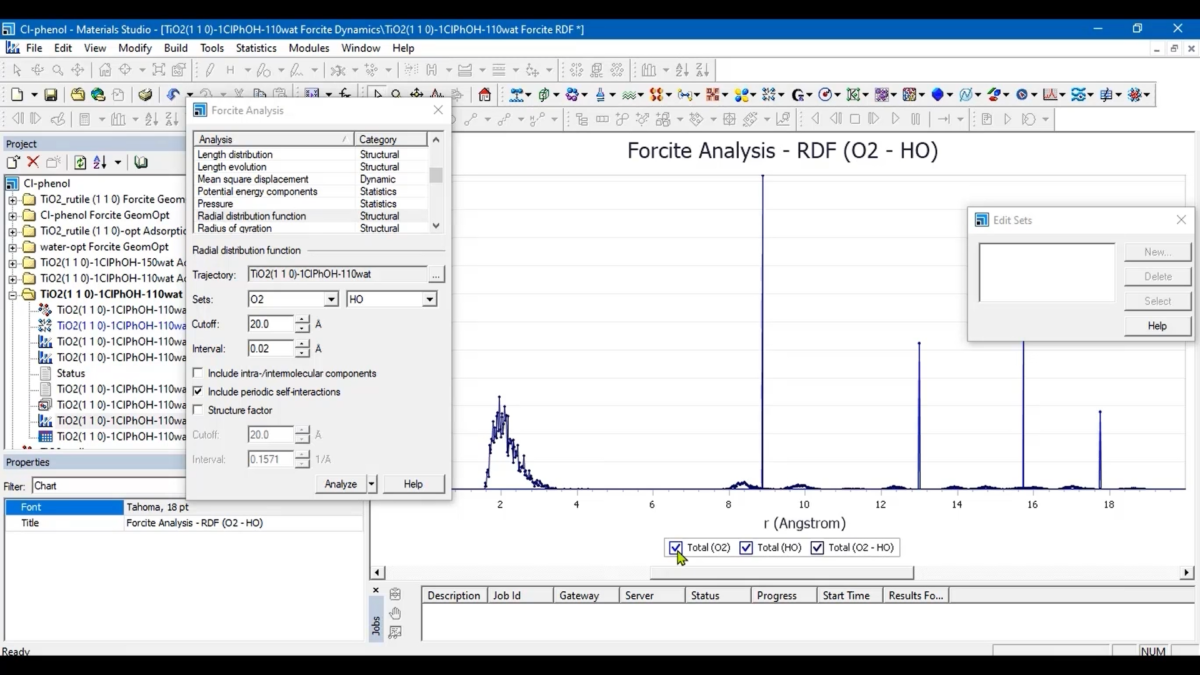
超详细的-使用Materials Studio软件进行径向分布函数(Radial Distribution Function,RDF)分析
一、理论简介:
径向分布函数(Radial distribution function, RDF)是一个在凝聚态物理中频繁使用的参数,它描绘了粒子密度如何随着距离的变化而变化。具体来说,RDF是在r到r+dr之间的平均粒子密度比上总平均的粒子密度。换句话说,如果我们有一个参考粒子并想知道其他粒子在空间中的分布几率(离给定粒子多远),那么我们可以查看RDF。

对于紧密排列的系统,RDF会先快速达到峰值,然后降低到1。这是因为当粒子之间的距离减小时,相互之间的影响也会增大,从而使得粒子密度增大。然而,当粒子之间的距离达到一定程度后,相互之间的影响开始减弱,因此粒子密度也会随之减小。
在实际应用中,RDF具有广泛的应用价值。例如,通过分析径向函数,我们可以判断每个原子在某个半径内所有受到作用力的情况。这在分子模拟中尤其有用,因为分子间存在的相互作用力是研究的重点。此外,径向分布函数还可以描述聚合物链在空间中的延伸,是表征链构象的重要参数之一。

图1 径向分布函数概述图
二、模块简介:
那么在Materials Studio软件中怎么操作呢?下面与东小软一起Look一下:在Materials Studio软件中,有两大模块可以直接进行径向分布函数的分析,首当其冲的是分子力学领域的核心模块-Forcite Plus模块,这是一个先进的经典分子力学工具,含有一组全面的包括从简单分子到二维表面到晶体这样的分析工具使模拟人员可以分析类似密度变化这样的简单性质到计算偶极相关函数这样的复杂性质。

三、操作简介:
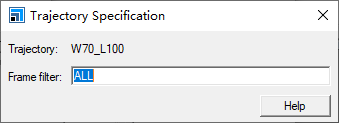
Forcite Plus分析使用1 Å3的参考体积。为了比较有无周期性边界条件的模拟,后者得到的g(r)必须乘以前者使用的单元格体积(以Å3为单位)。随着r的增大,g(r)趋向于1。Structure/Trajectory:一个有效的3D原子文档(.xsd)或一个3D原子轨迹文档(.xtd),这个文档的文件名将会显示在文本框中。如果想选择当前活动的轨迹文档的特定帧进行分析,点击按钮以访问轨迹规范对话框。默认情况下,除非指定特定帧,否则当前活动的轨迹文档中的所有帧都将包含在分析中。

这个控件的名称会随着当前范围内的文档的性质而改变。
Sets&Cutoff:在Forcite Plus模块中,径向分布函数可以针对一个或两个原子集合进行计算。在单一集合的情况下,该函数会计算集合中所有原子或质心的配对,这些配对的距离小于截止值。对于两个集合,该函数会计算每个独立集合内的所有原子或质心的配对,以及包含来自每个集合的一个原子或质心的所有配对,忽略所有距离大于截止值的配对。

Interval:指定径向分布函数计算的bin宽度,单位为埃。由于径向分布函数是作为直方图计算的,较小的bin宽度会导致更多的bin和更精细的分辨率图。然而,当bin的数量接近数据点的数量时,图的分辨率将会降低。

Include intra-/intermolecular components:在执行径向分布函数计算时,Forcite允许我们选择生成径向分布函数的分子内和分子间组分的单独图表,除了总的径向分布函数。

Include periodic self-interactions:包括周期性自我相互作用:当选中时,表示将在计算中包括原子或质心与其周期图像之间的相互作用。默认值 = 选中。如果截止距离大于最短的单元向量,并且在计算中包括周期性的自我相互作用,径向分布函数可能会显示出与单元向量长度相对应的显著峰值。

Structure factor:我们还可以选择在运行过程中计算结构因子。结构因子是从径向分布函数的傅立叶变换中得出的,也可以通过X射线和中子衍射实验测量,这为与模拟进行比较提供了有用的信息。

设置径向分布函数计算的步骤如下:
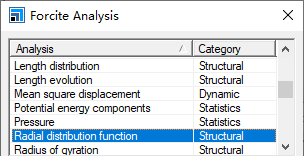
1. 将经过几何优化与动力学弛豫后的稳定结构至于当前,从菜单栏选择Modules | Forcite | Analysis,以显示Forcite分析对话框。

2. 从对话框顶部的网格中选择径向分布函数(Radial distribution function)。
3. 确保要分析的3D结构文档是当前文件。文件的名称将出现在Structure/Trajectory文本框中。
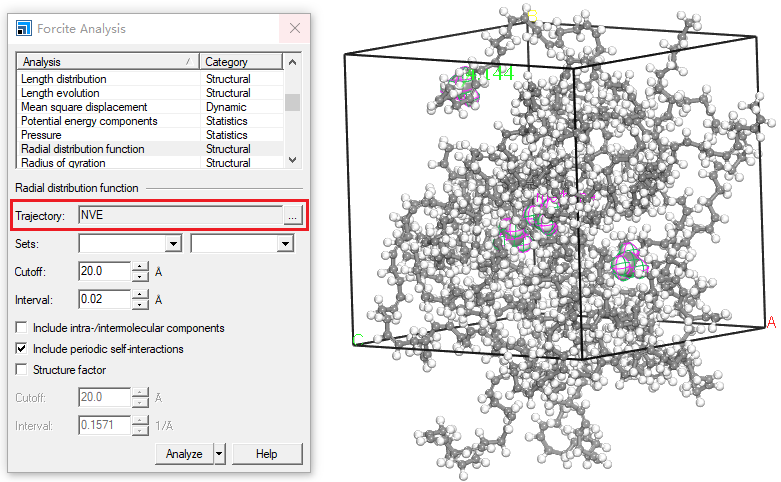
4. 如果希望指定来自轨迹文档的特定帧进行分析,点击按钮以显示轨迹规范对话框,并在Frame filter文本框中输入适当的帧编号。

5. 如果需要,将计算限制为一或两个特定的原子组,可以通过从集合下拉列表中选择预定义的集合,或者通过在结构中手动选择希望包含在计算中的原子来实现。

6. 手动选择原子只允许定义一个集合。如果希望计算两组原子的径向分布函数,你需要在开始分析之前定义集合。这最好是使用原子选择或查找模式工具来完成的。
7. 指定原子间Cutoff以限制参与计算的原子对的数量(一般cutoff设置为结构三边最短的一半),并设置Interval以调整径向分布函数图的分辨率。

8. 如果需要径向分布函数的分子内和分子间组分的单独图表,除了总的径向分布函数,请勾选包括分子内/分子间组分复选框。

9. 如果希望在运行过程中计算结构因子,请勾选结构因子复选框。为结构因子计算指定原子间截止距离和频率间隔。

10. 点击Analyze按钮。结果分析:
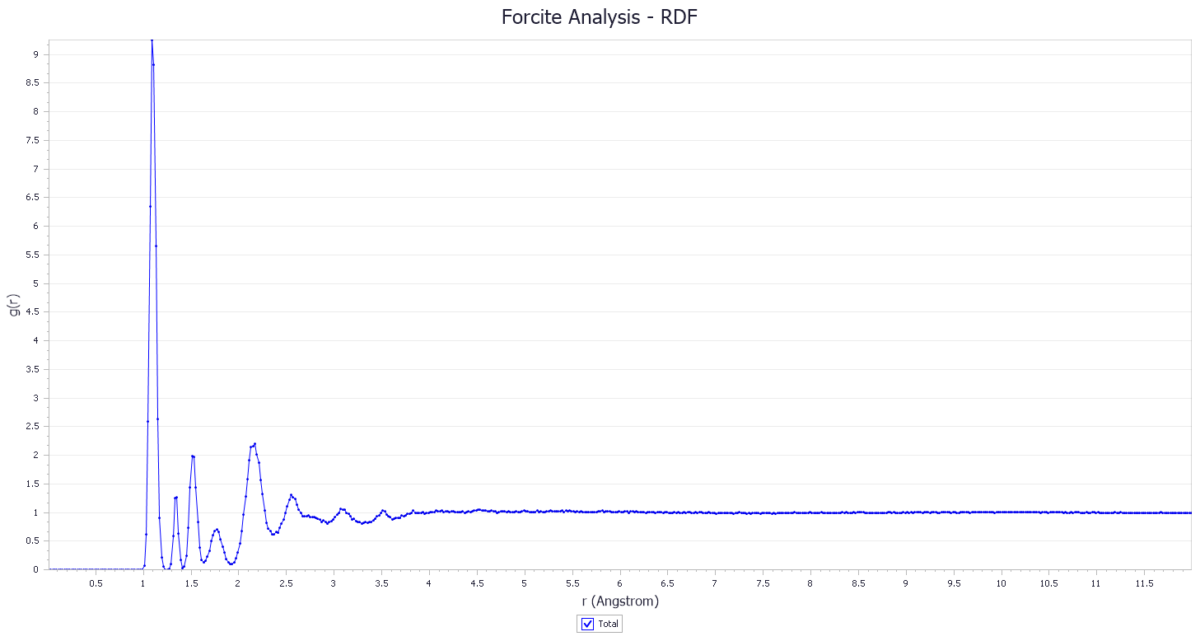
峰值:径向分布函数图的峰值表示在某个特定距离上出现原子的可能性。峰值的位置可以反映出原子间的平均距离。
峰宽:峰宽反映了原子间距离的分布范围。峰宽越大,表示原子间距离的分布范围越广。
峰高:峰高反映了在某个距离上出现原子的概率。峰高越高,表示在该距离上出现原子的概率越大。
峰的数量和分布:峰的数量和分布可以反映出体系的结构特性。例如,常见的晶态结构中每个原子的位置都是固定的,是有序的结构,其径向分布函数具有长程的峰,而对于非晶结构,其径向分布函数一般只有短程的峰。
四、视频教程
别着急,上述只是文字操作,如果想要更像生动形象的视频,东小软也悉心为您准备啦,点击以下链接可以直接观看~~~~
【Materials Studio Forcite模块-RDF径向分布函数分析】 https://www.bilibili.com/video/BV1EN411A7LG/?share_source=copy_web&vd_source=0795c81567f7e76676a818259a8ffcda -
 2023-11-16 09:06:21
2023-11-16 09:06:21要在 Materials Studio 中使用径向分布函数(RDF)分析耗散粒子动力学(DPD)模拟胶束自组装中胶束质心的 RDF,你可以遵循以下步骤:
步骤一:准备模拟数据
运行 DPD 模拟:使用 Materials Studio 中的 DPD 模块来模拟胶束自组装过程。
收集数据:确保模拟运行完成后,得到所需的数据文件,包括胶束质心的轨迹信息。
步骤二:打开 Materials Studio
打开分析工具:启动 Materials Studio,并选择用于分析的适当模块(如 Forcite 或其他分子动力学分析工具)。
加载数据:导入模拟输出的数据文件,以便进行后续的分析操作。
步骤三:计算径向分布函数(RDF)
选择质心坐标:从加载的数据中选择胶束质心的坐标数据。
计算 RDF:使用 Materials Studio 中的分析工具来计算胶束质心之间的 RDF。选择两个胶束质心之间的距离作为关注的对象。
使用 RDF 工具计算所选胶束质心之间的距离分布。
步骤四:分析结果
解释 RDF 曲线:分析得到的 RDF 曲线,观察峰值位置和形状。
关联胶束结构:RDF 曲线的峰值和形状可提供关于胶束结构和组装方式的信息。例如,峰值位置可以指示胶束之间的平均距离,峰值的形状和数量可以反映胶束结构的对称性和密度分布。
注意事项:
在计算 RDF 时,确保选择合适的分析参数和模拟数据,以获得准确和可靠的结果。
对于特定的系统和模拟条件,可能需要调整分析设置和参数来获得最具信息量的 RDF 结果。
Materials Studio 提供了详细的文档和教程,可以帮助你更深入地了解如何使用其分析工具进行 RDF 分析,并优化分析步骤以适应你的研究需求。
请注意,具体操作步骤可能因版本或软件更新而略有不同。如果你在操作过程中遇到困难,建议查阅 Materials Studio 的官方文档或寻求专业支持以获取更详细的指导。
回答的内容不能为空
还能输入100个字符 -
-
materials studio中构建的模型,图像消失了,怎么解决?
-
2023-11-28 10:21:51
对于MS中图像显示的问题,在确定不是其他硬件方面(电脑自身)的原因的前提下,可以按照以下步骤方法:
1. 打开MS(Materials Studio)界面。
2. 点击工具(tools)。
3. 选择选项(options)。4. 点击图形(Graphics)。
5. 检查其中的选项是否已勾选。
6. 如果未勾选,请全部勾选。
7. 重新启动MS。
8. 如果已经勾选,请全部取消勾选。
9. 重新启动MS。
通常情况下,上述方法可以解决大部分MS软件中图像显示的问题。 -
 2023-11-15 14:04:03对于MS中图像显示的问题,都可以尝试: MS界面→tools→options→Graphics :查看其中的选项是否勾选,如果没有勾选,全部勾选,重启MS;如果已经勾选,现全部不勾选,重启MS。
2023-11-15 14:04:03对于MS中图像显示的问题,都可以尝试: MS界面→tools→options→Graphics :查看其中的选项是否勾选,如果没有勾选,全部勾选,重启MS;如果已经勾选,现全部不勾选,重启MS。
回答的内容不能为空
还能输入100个字符 -
-
Materials Studio为了构建某晶体的晶胞结构,需要知道哪些信息?
-
2023-11-28 10:27:21
在Materials Studio中,构建晶体的晶胞结构是一项关键任务。为了成功完成这一任务,您需要掌握以下三个重要信息:空间群、晶格常数和原子坐标。
首先,让我们来了解一下空间群。空间群是描述晶体对称性的一个重要参数。它决定了晶体中各个原子的位置关系以及晶体的整体形状。以225号空间群为例,由于要求a=b=c,α=β=γ=90°,实际上只需要调整一个参数即可。这意味着在这个空间群中,晶体的三个边长相等,且三个角度均为90度。通过选择合适的空间群,您可以确保晶体的结构符合预期。
其次,我们需要了解晶格常数。晶格常数包括晶胞尺寸和夹角。对于Ag晶体来说,我们可以将晶格常数设定为4.0857Å。这个值决定了晶体中原子之间的距离和相对位置。通过准确设置晶格常数,我们可以确保晶体的结构与实际材料相符。
最后,我们需要确定原子坐标。原子坐标描述了晶体中各个原子的位置信息。以ZrO2晶体为例,我们可以在XYZ坐标(0.25,0.75,0.25)的位置添加O原子,同时在坐标(0,0,0)的位置添加Zr原子。通过准确指定原子坐标,我们可以确保晶体中的原子按照预期排列。
综上所述,有了这三个关键信息——空间群、晶格常数和原子坐标,您就可以在Materials Studio中成功构建晶体的晶胞结构了。这些信息的准确性和完整性对于获得可靠的模拟结果至关重要。因此,在进行晶胞结构构建时,务必仔细考虑并正确输入这些信息,以确保您的研究工作能够顺利进行并取得理想的结果。
-
2023-11-15 14:02:23构建某晶体的晶胞,以下信息缺一不可: 第一,该晶体晶胞的空间群,这是晶胞的对称性信息; 第二,该晶体晶胞的晶格常数,一共包括6个参数,其中长度参数3个:a、b、c;角度参数3个:α、β、γ; 第三,该晶体晶胞中所有原子的三维空间坐标(x, y, z)。
回答的内容不能为空
还能输入100个字符 -
-
什么是Pipeline Pilot?
-
2023-11-28 10:28:47
BIOVIA Pipeline Pilot是一款科学数据分析和工作流程管理软件,由美国BIOVIA公司开发,广泛应用于实验室信息学中。它不仅用于企业级软件实施,也服务于不同软件之间的数据传递,帮助用户解决系统集成的挑战。
在功能上,Pipeline Pilot为用户提供一系列“标准化”,“结构化”的数据处理组件,可实现各种格式数据提取,数据查询,数据转换、数据分析,以及定制化分析报告等。从文本挖掘、化学信息学、ADMET到序列分析和报告,Pipeline Pilot提供了一组丰富的“开箱即用”的工具包来实现常见的可重复使用的工作流任务。例如,你可以使用Pipeline Pilot提供的算法组件,或者使用开源的科学数据处理和转换算法,在Pipeline Pilot平台构建出工作流的自动化执行任务,实现基因序列的子结构或者相似性搜索,读取基因组数据或者孔板数据集,使用复杂的算法分析图像,对齐基因序列数据,修剪短片段或长片段基因系列,评估运行质量。
此外,基于BIOVIA Pipeline Pilot构建的Discovery Studio模块,为实验生物学家、药物化学家、结构生物学家、计算生物学家和计算化学家等提供综合的分子模拟平台。用户可以像组装电子产品一样,使用Pipeline Pilot中的元器件自定义工具,满足各类特定需求。通过优化研究创新周期、提高工作效率以及减少研究和IT经费,Pipeline Pilot能够发挥巨大的作用。
-
2023-11-15 11:11:40Pipeline Pilot(PP) 的是 Accelrys 公司旗下的—款图形化工作流软件,具 备功能强大的信息整合和流程定制能力, 其在优化研究创新周期、提高工作效率与减少研究和IT
回答的内容不能为空
还能输入100个字符 -
-
材料基因组项目中分子模拟能做什么?
-
2023-11-28 10:31:09
在材料基因组项目中,分子模拟起着至关重要的作用。科学家可以利用分子模拟找出元素间的相互作用对材料的种类和性质带来的广泛影响,以此为基础,期望以更短的周期为不同应用“定制”相应材料。
分子模拟的运行可以分为两个方面:一是依据实验数据推测研究对象的三维结构和理化性质;二是利用第一性原理方法进行尽可能准确的预测,如量子化学方法和力场计算方法。
此外,随着材料基因组计划(MGI)的提出以及大数据技术和人工智能技术的飞速发展,基于数据驱动的新材料设计得到了广泛的关注,并逐渐成为新型材料研发的重要方法。例如,通过机器学习等技术加速实现由传统的实验驱动、理论驱动向数据驱动、智能模型驱动创新模式的转变。
值得一提的是,王院士(王崇愚,我国材料基因组计划的积极倡导人和推动者之一)向我们介绍了材料基因组中高通量实验表征的思路,一是指基本单元经过高通量合成、表征与筛查,经过基本单元试样模拟合成和基本单元高通量表征筛查;另一方面指基本单元组装过程及介观尺寸新材料高通量表征功能,针对不同新材料具备基本单元组装试验模拟功能,组装过程原位表征功能,再组装介观材料大尺度统计分布高通量表征功能。这种大规模的并行运算和模拟可以极大地提高新材料的研发效率。
-
2023-11-15 11:10:35科学家想通过Materials Genome Initiative(MGI)项目,找出元素间的相互作用对材料的种类和性质带来的广泛影响,以这些知识为基础,希望以更短的周期为不同应用“定制”相应材料。
回答的内容不能为空
还能输入100个字符 -



 苏公网安备 32059002002276号
苏公网安备 32059002002276号
功能:
性价比:
易用性: