




所需功能和模块:Discovery Studio Client, DS CDOCKER.
所需数据文件:1EQG.dsv, 1EQD-ibuprofen-conf.sd, 1EQG-ibuprofen.sd
所需时间:15分钟
介绍
CDOCKER是基于CHARMm力场的分子对接方法,这种方法可以产生高精度的对接结果。在本教程中,天然布洛芬配体分子对接回COX-1受体的结合位点中,得到的对接构象和X-ray衍射得到的晶体结构中的配体天然构象进行比较。本教程包括:
准备对接体系
运行CDOCKER
CDOCKER结果分析
计算对接结果的RMSD值
准备对接体系
在文件浏览器(Files Explorer)中,找到并双击打开Samples | Tutorials | Receptor-Ligand Interactions| 1EQG.dsv。
在分子窗口中将打开一个带有活性位点的蛋白质三维结构(图1)。
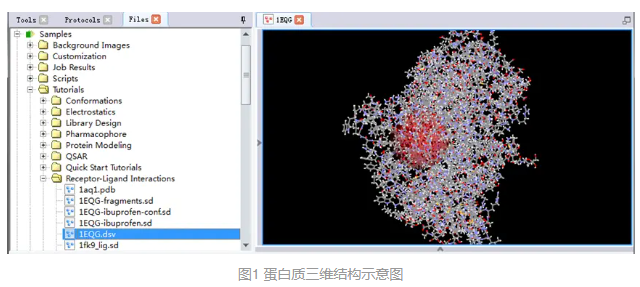
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Define and Edit Binding Site,依次点击Show/Hide Residues Outside Sphere和Show/Hide Sphere。
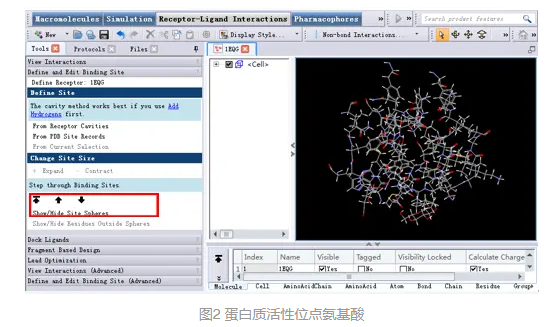
展开菜单栏View|Transform,点击Fit To Screen将结合位点的氨基酸在窗口中居中显示(图2)。
以上操作可以将结合位点外的残基以及球体隐藏,以便观察对接结果时更加便捷。
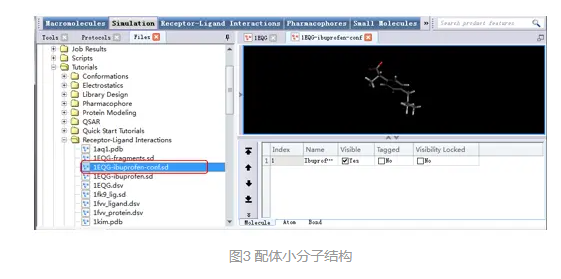
找到并双击打开Samples | Tutorials | Receptor-Ligand Interactions| 1EQG-ibuprofen-conf.sd文件。
将打开一个具有随机构象的布洛芬分子(图3)。
运行CDKCKER
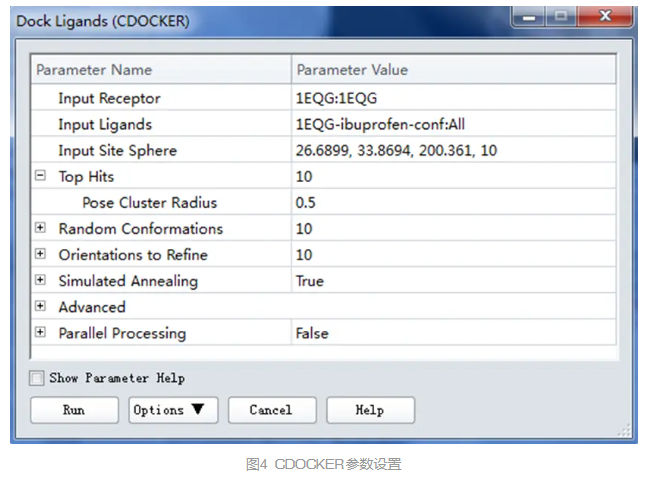
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Dock Ligands,点击Dock Ligands (CDOCKER),打开相应参数面板。
在参数面板中,将Input Receptor设置为1EQG:1EQG。
参数Input Ligands设置为1EQG-ibuprofen-conf:All。
点击Input Site Sphere参数,从下拉列表中选择该sphere的坐标及半径。
展开Top Hits参数,设置Pose Cluster Radius为0.5。
将RMSD阈值设为0.5埃以确保对接构象尽可能具有多样性。
其余参数默认(图4),点击Run运行。
CDOCKER结果分析
作业完成后,在作业浏览器(Jobs Explorer)中,双击完成的分子对接任务,打开Report报告界面,点击View Results,打开对接结果。
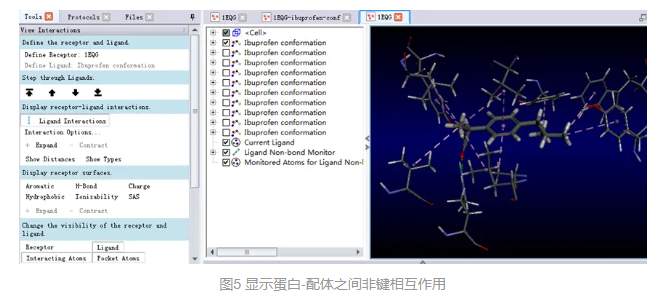
1.非键相互作用的直观显示与分析
在工具浏览器(Tools Explorers)中,展开Receptor-Ligand Interactions | View Interactions,点击Ligand Interactions。
在视图窗口中,受体蛋白氨基酸残基与配体对接poses间的非键相互作用会通过不同颜色的虚线显示出来,且只有参与了同配体之间的相互作用的残基才会显示。(图5)
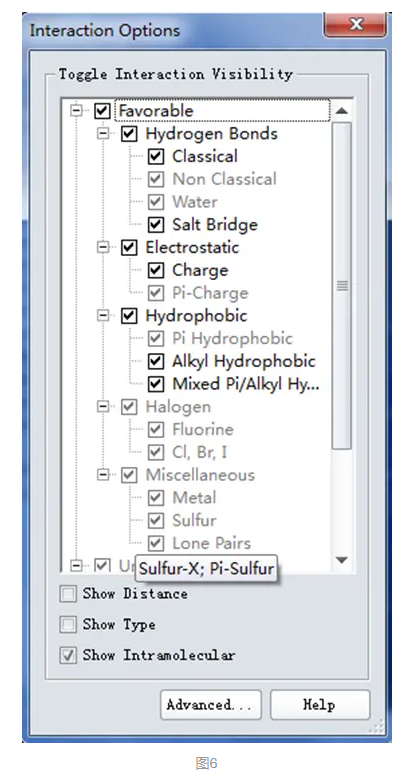
点击上述View Interaction工具面板下的Interaction Options,展开如下窗口(图6)。
该窗口中列出DS中所有非键相互作用类型,其中黑色显示表示该蛋白和配体间存在的非键相互作用。
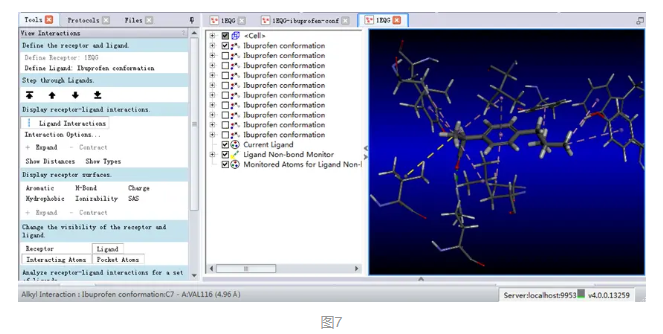
此外,在分子显示窗口任意选中某一虚线,在DS界面的左下方就会显示该非键相互作用类型及距离等相关信息。(图7)
为了更好的观察受体分子与配体对接pose间的相互作用,可以对体系进行旋转以获得更佳的观赏角度。
点击上述View Interaction工具面板下的
为了更好的观察受体分子与配体对接pose间的相互作用,可以对体系进行旋转以获得更佳的观赏角度。
点击上述View Interaction工具面板下的
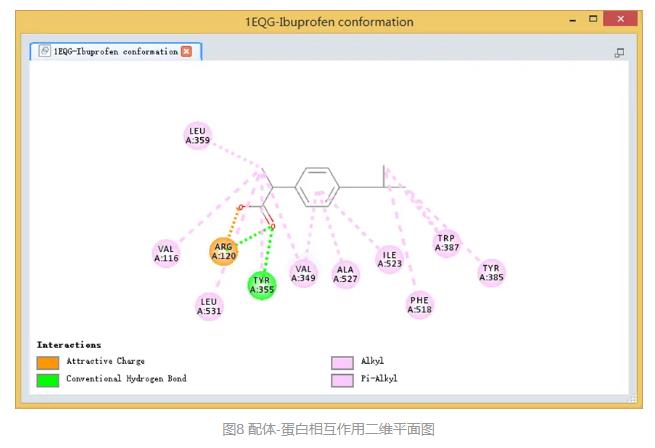
可以看到在一新窗口中打开配体-蛋白相互作用二维平面图,便于我们更直观的观察两者的相互作用及关键的氨基酸和基团。(图8)
3.对接配体和布洛芬天然晶体结构比对
在任务浏览器(Jobs Explorer)中,单击该任务条下(点开前面的+号)Docked Ligands链接。
对接配体将在一个新的分子窗口中打开。
按住CTRL+G,显示分子图形窗口。
展开菜单栏File | Insert From,点击File…,选择Samples | Tutorials | Receptor-Ligand Interactions | 1EQG-ibuprofen.sd。
在同一窗口中插入布洛芬的天然晶体结构1EQG-ibuprofen.sd。
在表格视图中,设置最后一行的Ibuprofen分子的Visible和Visibility Locked为True。
按住CTRL+Down,观察每个配体文件和布洛芬天然晶体结构的比对情况。
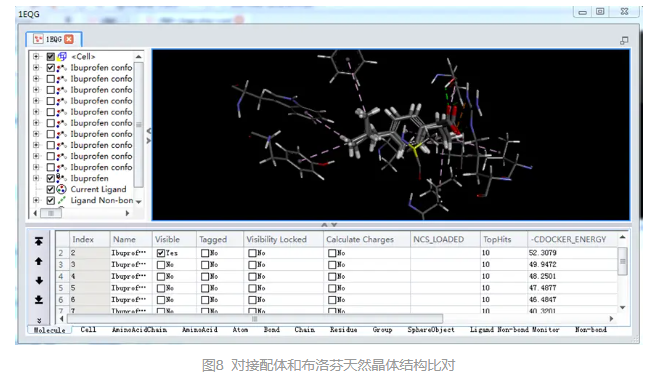
可以发现CDOCKER打分最高的位点,即-CDOCKER_ENERGY的值最高,和天然布洛芬晶体结构具有很好的叠合效果。
在表格视图中,选择最后一行的Ibuprofen分子,展开菜单栏Structure|RMSD,点击Set Reference。
在分子窗口中点击鼠标右键,选择Show All,显示所有小分子结构。
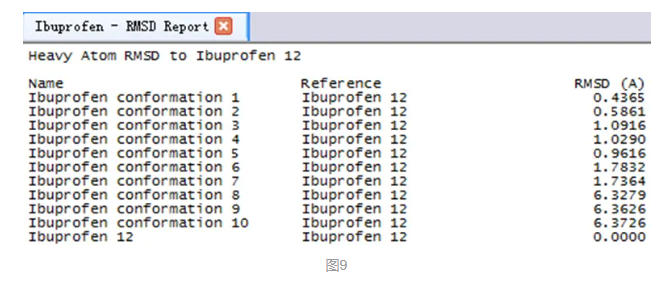
展开菜单栏Structure|RMSD,点击Heavy Atoms。
打开一个新的窗口,显示所有10个对接构象同晶体构象之间的RMSD偏差。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号