




目的:采用Libdock,以一组配体及一个明确活性位点的蛋白质为实例,示范分子对接及结果分析的操作过程。
所需功能和模块:Discovery Studio Visualizer client, DS LibDock, DS Catalyst Conformation。
所需数据文件:1kim_prot.dsv和kinase_ligands.sd。
所需时间:15分钟
介绍
基于结构的药物设计技术在药物研发中起着非常重要的作用。在药物分子产生药效反应的过程中,药物分子要与靶标相互结合,首先需要两个分子充分接近,采取合适的取向,使两者在必要的部位相互契合,发生相互作用,继而通过适当的构象调整,才能得到一个稳定的复合物构象。
基于结构的药物设计主要采用的是分子对接技术。分子对接就是把配体分子放在受体活性位点的位置,然后按照几何互补、能量互补以及化学环境互补的原则来实时评价配体与受体相互作用的好坏,并找到两个分子之间更佳的结合模式。分子对接是从整体上考虑配体与受体结合的效果,能够较好避免其他方法中容易出现的局部作用较好而整体结合欠佳的情况。在药物设计中,分子对接方法主要用来从小分子数据库中搜寻与受体生物大分子有较好亲和力的小分子,进行药理测试,从中发现新的先导化合物;或者用于解释药靶之间的作用机制,并在得到作用模式的基础上指导化合物结构改造。
LibDock是Discovery Studio中的其中一种对接方法。该对接方法首先会针对受体活性位点计算得到热区图,该热区图包含极性和非极性部分;接着不同构象的配体分子分别刚性地叠合至热区图以形成比较合适的相互作用;然后进行能量优化;最后保留打分较高的对接构象。
本教程将采用LibDock将一组配体分子对接到胸苷激酶(thymidine kinase)中,包括:
准备分子对接体系
执行分子对接计算
分析配体对接结果
准备分子对接体系
在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions| 1kim_prot.dsv文件。
该蛋白将在一个新的分子窗口中出现,该蛋白已经预处理过,且活性位点也已定义好。
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Define and Edit Binding Site,依次点击Show/Hide Residues Outside Sphere和Show/Hide Sphere。
展开菜单栏View|Transform,点击Fit To Screen将蛋白结合位点居中显示。(图1)
以上操作可以将结合位点外的残基以及球体隐藏,以便观察对接结果时更加便捷。
在文件浏览器(Files Explorer)中,找到并双击打开Samples| Tutorials| Receptor-Ligand Interactions| kinase_ligands.sd文件(图2)。
在新的分子窗口中打开一张共有9行的表格,每一行代表一个分子,共有9个配体分子。

执行分子对接计算
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interaction | Dock Ligands ,点击Dock Ligands (LibDock)执行分子对接计算。
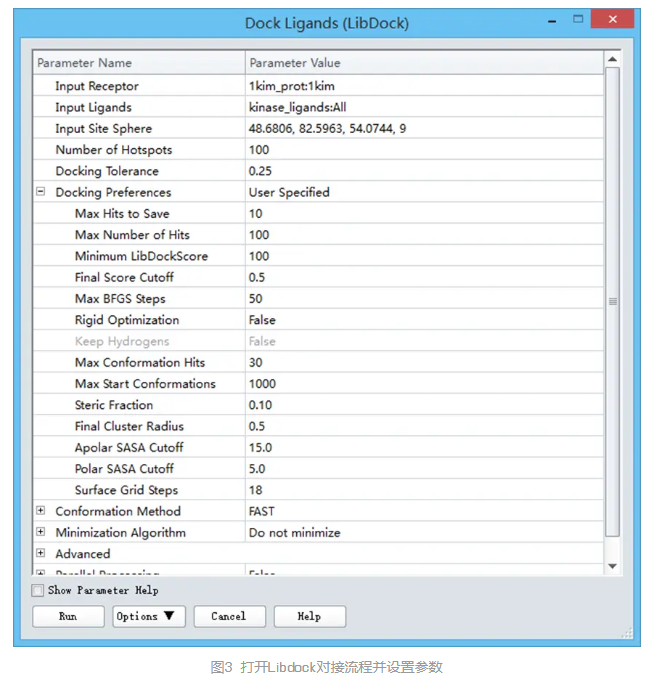
该模块相应参数在参数浏览器中打开。
在参数浏览器中,点击Input Receptor参数,从下拉列表中选择1kim_prot:1kim设置受体蛋白。
点击Input Ligands 参数,从下拉列表中选择kinase_ligands:All,指定对接配体。
点击Input Site Sphere参数,确保该参数设置为48.6808,82.5963,54.0744,9,如果不是该参数,则从下拉列表中选择该sphere的坐标及半径。
点击Docking Preferences参数,从下拉列表中选择User Specified,根据需要改变对接计算的特定参数。
展开Docking Preferences参数,点击Max Hits to Save参数,输入值10。
本教程中,修改默认设置以减少最终保留的对接构象,减少所用时间。(图3)
参数设置完毕后,点击Run运行作业。
分析分子对接结果
待作业完成后,对接结果会自动在一个新的窗口中打开,包含蛋白(只显示配体结合位点处残基)和所有对接构象。其中显示的蛋白已被锁在窗口中,当依次查看所有对接构象时该蛋白都可视。
(或者在作业浏览器(Jobs Explorer)中双击刚完成的分子对接作业,打开Report窗口,点击View Results,同样可以打开对接结果。)
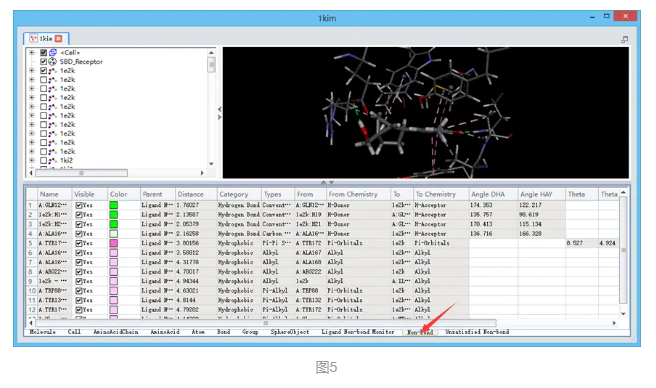
在打开的新的分子窗口1kim中确保系统视图(CTRL+H)和表格视图(CTRL+T)打开,在系统视图中可以观察到该窗口中包含了所有9个配体分子的对接结果,共81个。
点击表格视图中的

按钮,可观察排在第一位的对接构象,继而可通过点击表格视图中的

按钮(或者CTRL+Up/CTRL+Down),观察配体分子的每个pose同受体分子的结合模式。
1.非键相互作用的显示与分析
在工具浏览器(Tools Explorers)中,展开Receptor-Ligand Interactions | View Interactions,点击Ligand Interactions。
在视图窗口中,受体原子与配体对接poses间的非键相互作用会通过不同颜色的虚线显示出来,且只有参与了同配体之间相互作用的残基才会显示。(图4)

展开数据窗口,点击Non-bond,查看非键相互作用类型。
点击上述View Interaction工具面板下的
按钮可以观察不同的对接构象同蛋白之间的非键相互作用。
2.生成配体-蛋白相互作用二维平面图
选中并显示要描述的配体(如第一个1e2k),在工具浏览器(Tools Explorers)中,展开Receptor-Ligand Interactions |View Interactions,点击Define Ligand。
然后在该工具面板下,点击Show 2D Diagram。
可以看到在一新窗口中打开配体-蛋白相互作用二维平面图,便于我们更直观的观察两者的相互作用及关键的氨基酸和基团。(图7)

得到的二维相互作用图中各种图形显示元素的详细定义可参考图8:






 苏公网安备 32059002002276号
苏公网安备 32059002002276号