




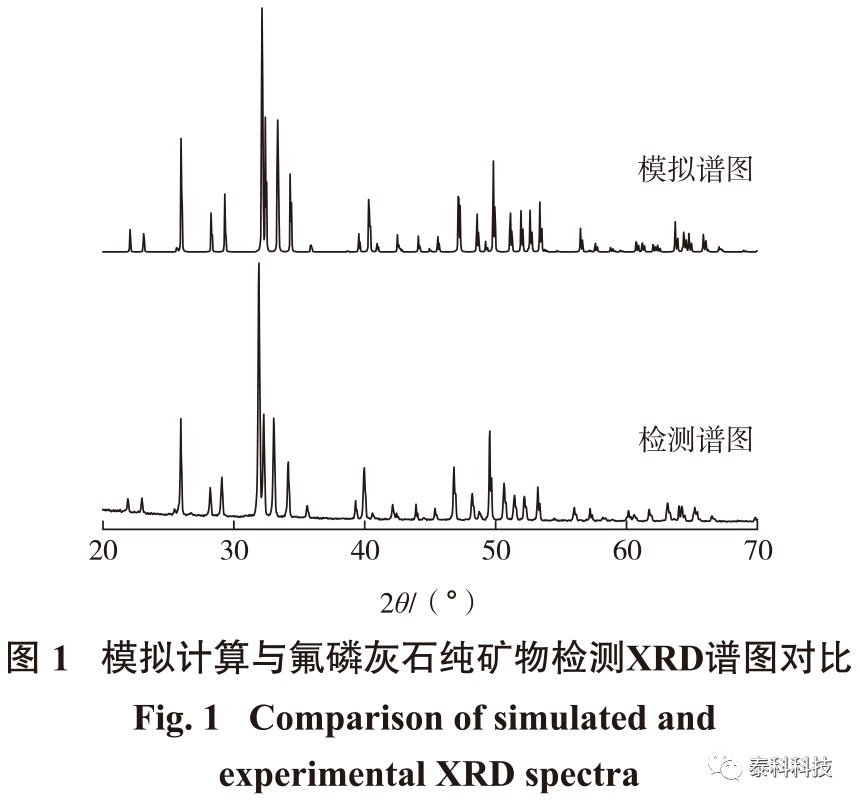
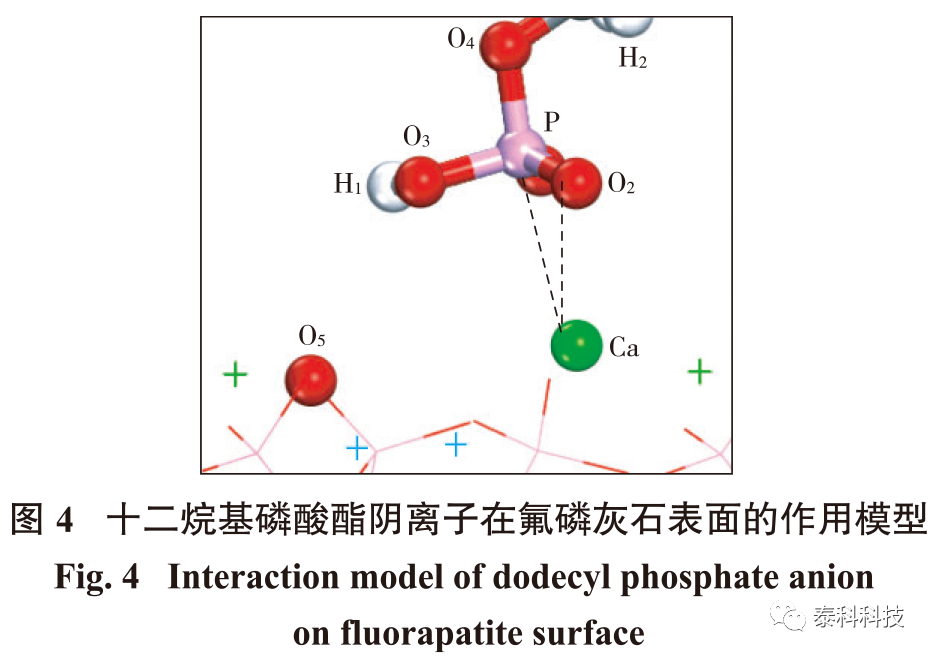
摘要:
为探究十二烷基磷酸酯分子结构对氟磷灰石浮选性能的影响,采用量子化学模拟手段,研究了十二烷基磷酸酯分子结构特性及其在氟磷灰石晶体表面的作用机理。采用Materials Studio软件的CASTEP模块,首先对分子态和离子态十二烷基磷酸酯的分子结构进行了优化,得到十二烷基磷酸酯分子及其阴离子的净电荷、Mulliken电荷布居、偶极距及最高占据轨道(HOMO)组成和能量。结果表明,相对于十二烷基磷酸酯分子,十二烷基磷酸酯阴离子有更强的供电子能力、范德华作用能力和反应活性。对十二烷基磷酸酯阴离子在氟磷灰石表面的相互作用模型进行了模拟计算,得到十二烷基磷酸酯与氟磷灰石之间的作用模型和吸附能,十二烷基磷酸酯阴离子与氟磷灰石表面之间的吸附能为负值,二者之间能够自发发生吸附作用,并形成化学键。通过单矿物浮选试验验证了模拟计算结果,即十二烷基磷酸酯可以作为氟磷灰石的浮选捕收剂。
引言:
在实际生产中,油酸是目前最常用的磷矿浮选分散性能不好、不耐低温等方面的缺点外,还有选择性差的问题。为了提高捕收剂在矿物表面吸附的选择性,科研人员从在理论和实践2个方面寻求突破。在理论研究方面,加拿大纽芬兰纪念大学的张亚辉通过总结现有浮选理论,提出了浮选药剂与矿物表面作用的“镜像对称规则”。该规则指出,矿物表面具有断裂键的金属离子倾向于与具备矿物晶体阴离子结构的浮选药剂发生作用。氟磷灰石分子式为Ca10(PO4)6F2,其晶体中具有磷氧四面体结构。按镜像对称规则,含有磷酸根或相近极性基团结构的分子易于同氟磷灰石表面的Ca离子发生相互作用。根据以上规则,十二烷基磷酸酯是氟磷灰石的潜在捕收剂之一。本文采用密度泛函理论对十二烷基磷酸酯的分子结构进行分析,并对十二烷基磷酸酯与氟磷灰石晶体表面的相互作用进行了模拟。

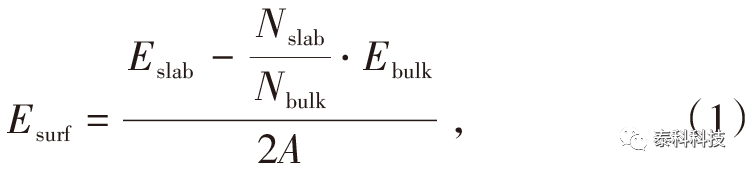












 苏公网安备 32059002002276号
苏公网安备 32059002002276号