



2022年第4期应用案例赏析-02
摘要
南京师范大学张义权教授团队采用全活空间自洽场(CASSCF)结合受限活性空间自旋相互作用和自旋轨道耦合(RASSI-SO)的方法研究了由镧系长链离子ErIII组成的单离子磁体(SIMs)为何不能具有大的能垒。该团队计算表明,由周围的等配位配体诱导的第一激发态克雷默斯双峰(KDs)中gx,y值越大,其横向磁矩越大,从而导致其第一激发态的快速量子隧穿磁化(QTMs)。当Er-L键长减小时,能垒没有不断增加。作者推断单核ErIII化合物不能通过增强周围的等配位场而拥有巨大的能垒。
引言
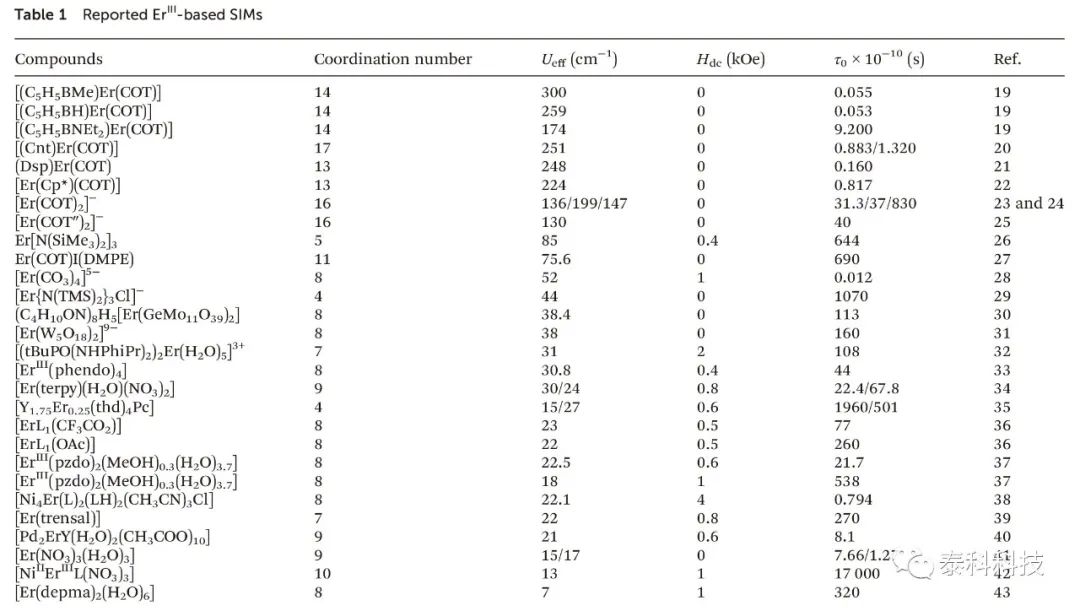
在过去的几年中,用于高密度数据存储设备的单离子磁体(SIMs)取得了迅速的进展,目前已报道了许多具有高能垒的高性能的离子磁体(SIMs)。作者在表1中列出了许多ErIII的化合物,其中[(C5H5BMe)Er(COT)]在基于ErIII-SIMs中具有更大的能垒,为300 cm -1。与ErIII-SIMs相比,SIMs与其它镧离子结合的性能更差。Yamashita等人报道了相似的分子结构Er(dbpc)3 和Er(btmsm)3 ,其能垒分别只有39.2和79.7 cm -1,远低于用DyIII组成的SIMs。为什么具有良性的配体环境的镧系离子ErIII组成的SIMs不能拥有DyIII-SIMs一样高的能垒? 南京师范大学张义权教授团队通过从头计算的方法来研究它们的磁结构寻找答案。
计算方法
南京师范大学张义权教授团队利用MOLCAS程序包对化合物1-3(图1)进行了基于单晶X射线测得的全活空间自洽场(CASSCF)计算。
结果讨论
化合物1-3的分子结构如图1所示,具有扁长的电子密度分布的中心离子ErIII与三个等配位的大体积配体耦合,从而为研究上述问题提供了机会。

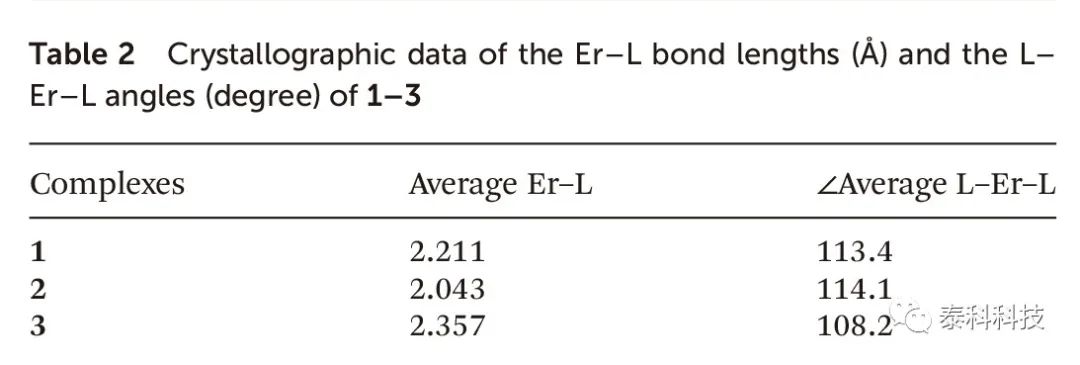
化合物1-3的晶体学数据如表2所示。
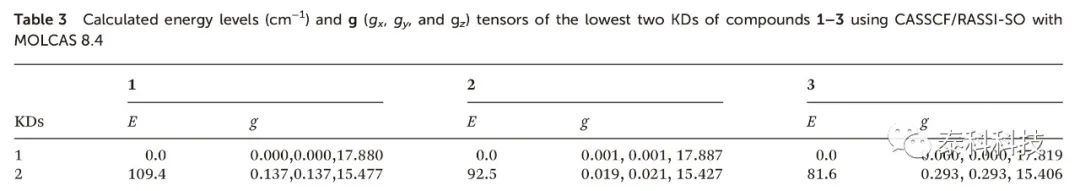
化合物1-3的克雷默斯双峰(KDs)的gx,y值均接近于零,而第一激发态KDs的gx,y值相对较大(表3)。
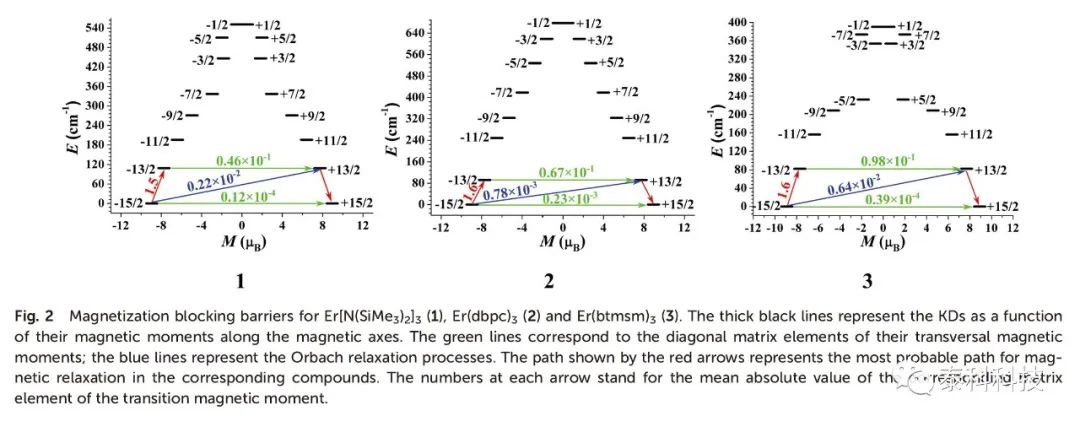
如图2所示,化合物1-3的横向磁矩分别为0.12×10-4、0.23×10-3和0.39×10-4μB,表明在低温条件下,其KDs中的QTMs可以被抑制。
如图3所示,通过观察三个分子结构,两个关键结构参数:(I)连接ErIII和L的矢量θ角(L = N (1), O(2)和C(3))与磁轴的夹角;(II)Er-L键的长度。
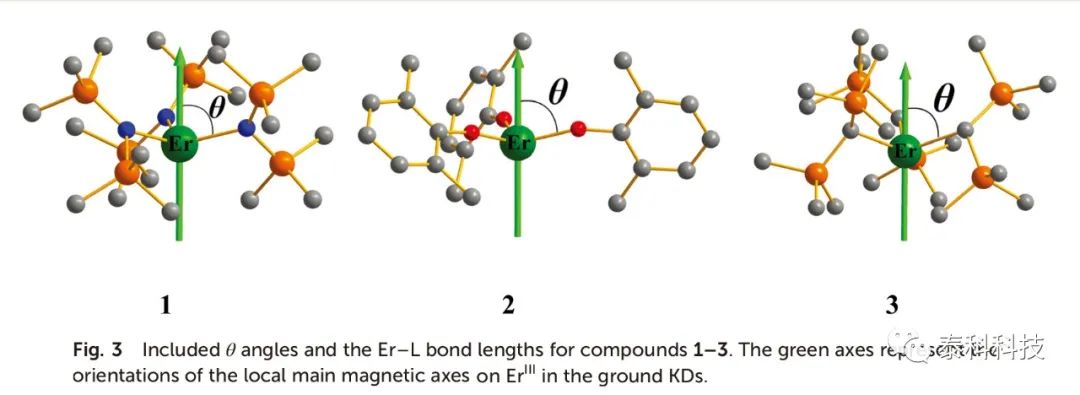
如表4所示,其中化合物1-3的θ角的平均值分别为74.8、75.7和69.3。
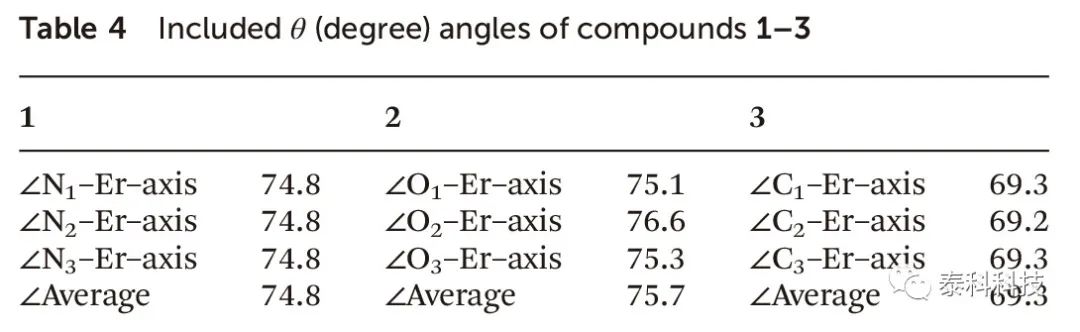
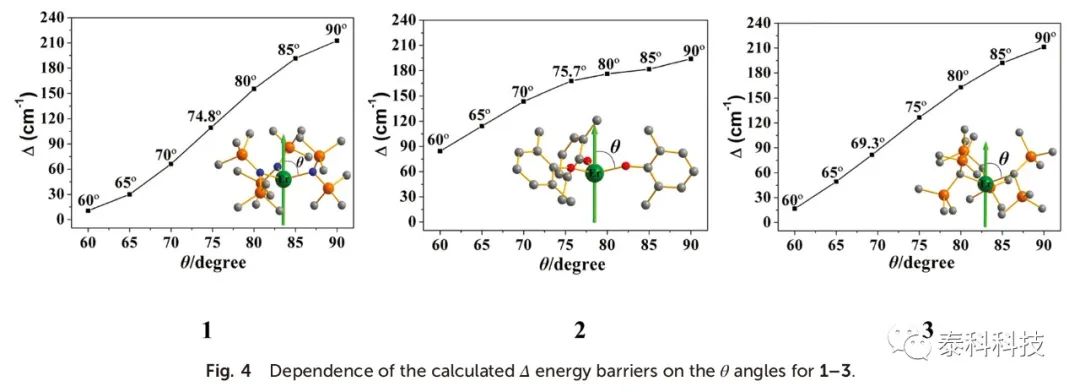
图4显示了计算出的Δ能垒对θ角的关系,其中所有的Δ随着θ角的增加而增加。
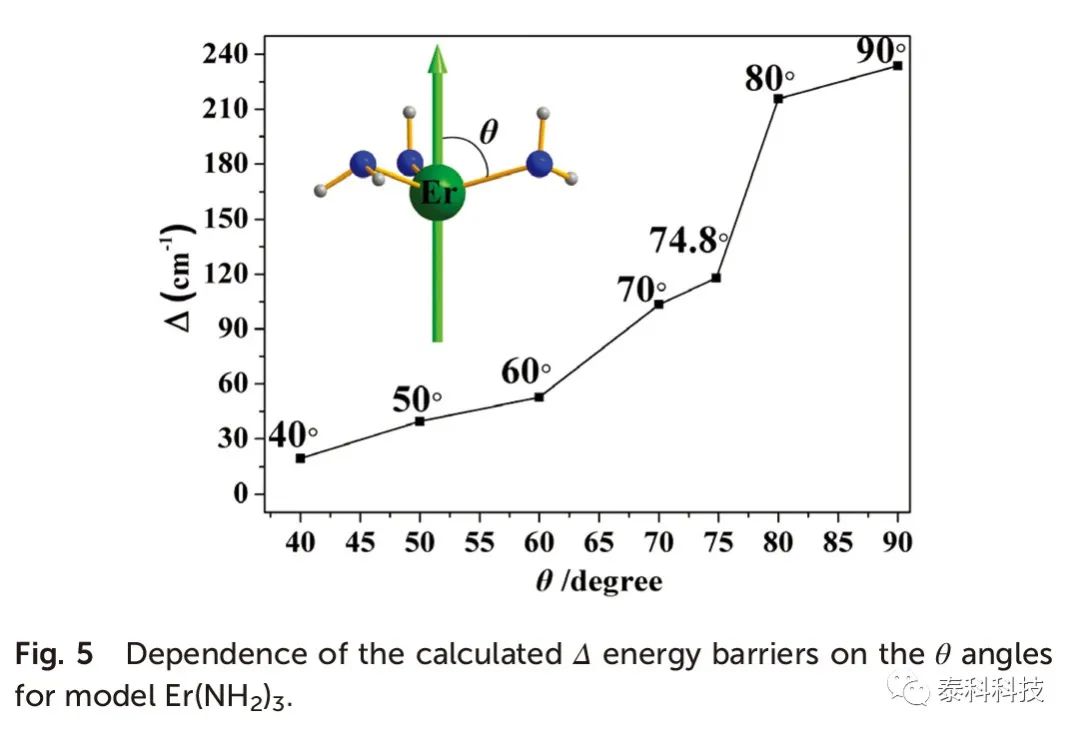
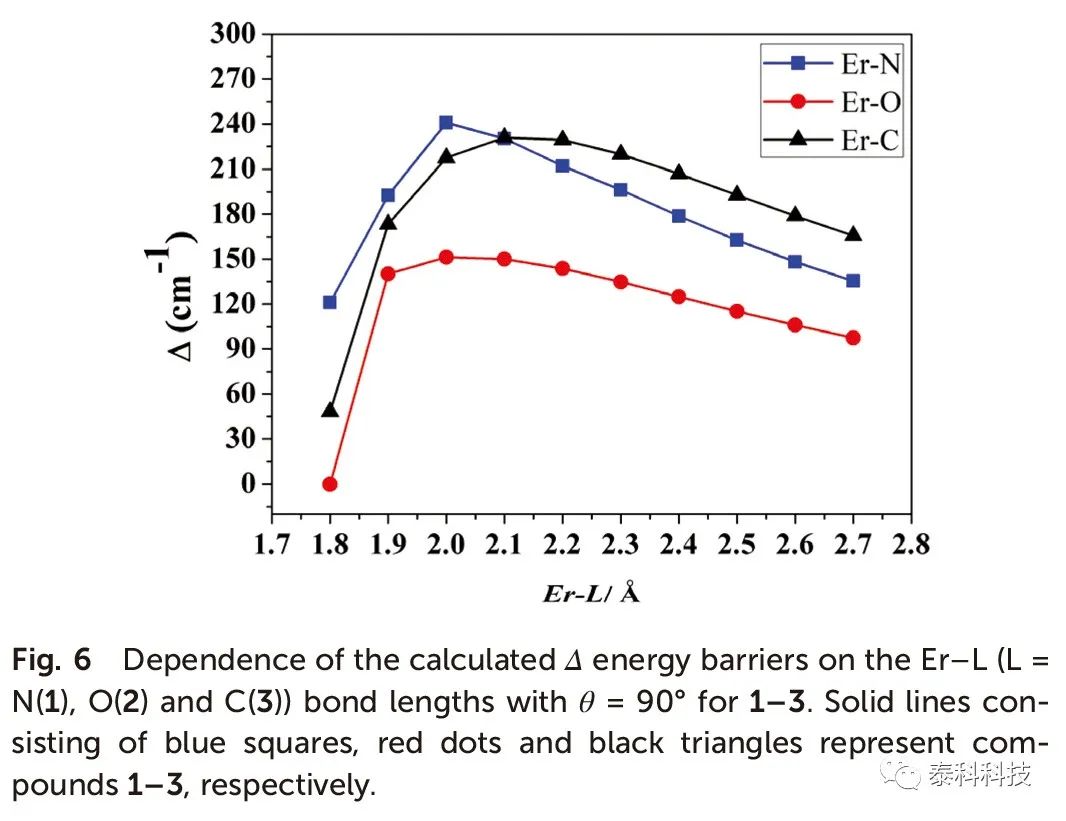
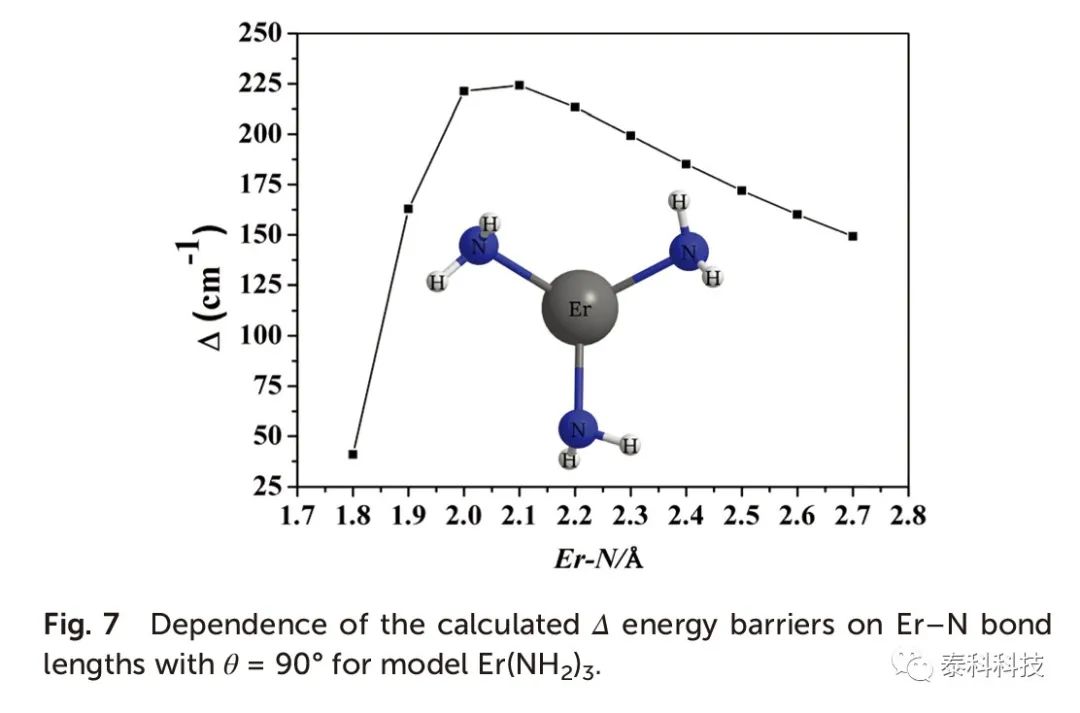
从图5可以看出,Δ能垒也随着θ的增加而增加,而θ= 90o时更大能垒仅为233.7 cm -1。当θ= 90o时,化合物1-3的Δ能垒与Er-L键长度的关系如图6所示。在θ= 90o时,计算得到的Δ能垒与Er-N键长的关系如图7所示,其趋势与图6相似。
总结
本文南京师范大学张义权教授团队采用CASSCF/ RASSII-SO方法,研究了为什么三个具有等配位配体环境的ErIII化合物不能通过增强周围的等配位配体场拥有像DyIII那样高的能垒。
(1) 计算出的能垒随θ的增大而增大,但三种化合物的更大能垒仅为212.6 cm -1。实际上,如果考虑离共振声子模和振动驱动QTM对能垒的影响,更大能垒将远低于212.6 cm-1。
(2) 当增加Er-L键距离时,化合物1-3的Δ值则减小了。但是,当Er-L键距离减小时,所有模型的Δ值都没有增加。
(3) 所有化合物和ErIII周围的等配位配体诱导的模型,其基态或第一激发态 KDs中的gx,y值都稍大一些,这将导致它们的横向磁矩更大,从而允许它们的基态或第一激发态KDs中存在快速QTMs。然而,随着Er – L键距离的减小,接近的ĤCF和ĤSO会导致基态和激发态KDs中几种mJ态的混合,从而产生较大的QTMs和较小的能级分裂。
综上所述,具有等配位配体环境的单核ErIII化合物很难通过增强周围的等配位配体场来获得像DyIII-SIMs那样高的能垒。
本文所有原子的基组是来自MOLCAS ANO-RCC的原子自然轨道。
作者简介
张义权 南京师范大学教授
江苏丹阳人, 中共党员, 2009年获南京师范大学博士学位,2012年北京大学化学与分子工程学院博士后。江苏省青蓝工程优秀青年骨干教师。主要研究方向为分子磁性材料的理论研究。
公司简介
北京泰科博思科技有限公司(Beijing Tech-Box S&T Co. Ltd.)成立于2007年,是国内领先的分子模拟及虚拟仿真综合解决方案提供商。
【更多MOLCAS应用实例请关注泰科科技公众号】





 苏公网安备 32059002002276号
苏公网安备 32059002002276号