


此白皮书调查了医疗器械风险管理的一些典型挑战, 并演示了如何使用 Siemens Digital Industries Software 解决方案显著改进流程。其解决方案提供坚实的资源库和工作流管理功能,可以简化需求管理和报告之类任务,同时使深层可追溯性自动化。
几乎从人类开始医疗以来,医疗器械产品开发就一直是医药执业和改进的基础要素。6,000 多年前,人们就开始使用研钵和研杵准备药粉。
在过去的这一千年里,医疗器械已经变得更加复杂和强大,法规环境也同样如此。法规的制定极为重要,因为它们可以帮助确保医疗器械在设计时经过验证与确认 (V&V)、符合所需的规范,并且可以用于预期临床用途。
从概念上而言,风险管理看似足够简单。由于潜在的危险情况可能会造成伤害,因此必须记录并减轻这些潜在的危险情况,从而控制结果并确保可以有预测性地、安全地使用医疗器械。
但是,每一位负责合规的人员都知道,忙中生乱,尤其是要处理多种法规、相互冲突的定义以及要在数字化级别追踪合规性的情况下。这种复杂性是由多种因素造成的,包括描述系统各组件相互之间关系所需的变量数量、选择让这些概念独一无二还是重复可用的选项、追踪合规性所需的多对多关系,以及时常模糊不清或令人费解的法规要求。
目前用于追踪这种复杂性的工具简单而不简约。尽管电子表格可以很好地追踪一对一关系,但在追踪一对多和多对多关系以确 保医疗器械产品开发合规性时,电子表格很快就难以满足要求。
电子表格和 Microsoft? Word 软件文档对于实现可追溯性的基础任务并不适用。可追溯性在设计和开发阶段是质量控制的核心, 在将监控报告关联回特定产品需求和验证与确认过程时,可追溯性仍然是验证与测试所必需的。
幸运的是,Siemens Digital Industries Software 为医疗器械产品开发提供了强大的解决方案。
从传统意义而言,开发人员面临的其中一项较为困难的系统开发任务就是实施有效的医疗器械风险管理。这里的关键词在于 “有效”,风险管理必须贯穿于从初始设计和制造到上市后监管,才 能成为整个过程必不可少的一部分。
大部分的困难来源于陈旧技术的使用,从静态手写文档和电子表格到追踪复杂动态环境,其中适用标准和其他产品需求会随着时间和设计开发迭代不断追踪。
医疗器械风险管理必须以正在使用的设计元素为基础,这些元素可以通过中央资料库与相关人员共享以供更新文档与保持版本控制。追踪设计单元关系时如果离开自动化,设计意图可能会在工作流程、产品更改和需求更新时丢失。
医疗器械产品开发工作是一项高度集成而遵守法规的过程。医疗器械风险管理中实施的两种主要标准为国家标准化组织 (ISO) 14971:2009 和 ISO 技术信息报告 (TIR) 24971:2013;前者为
制造商指定了识别医疗器械相关危险的过程,后者为实施风险管理过程中特定于 ISO 14971 的问题解决提供指导说明。欧洲国家还融合了 EN ISO 14971:2012,这一标准在几个重要方面不同于 ISO;如果哪个公司销售医疗器械到欧洲,就必须遵循此标准。
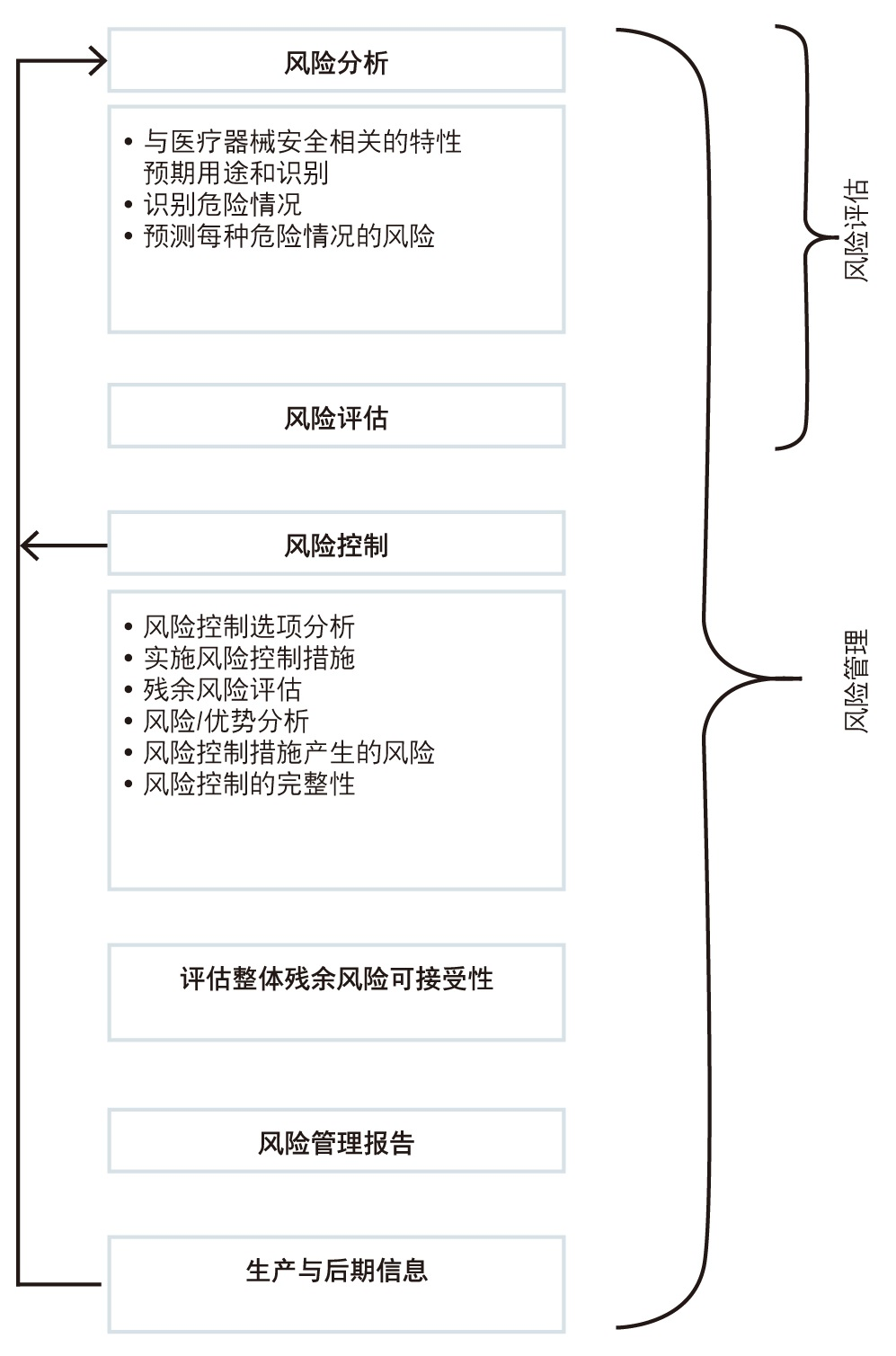
如图 1 所示,完整的医疗器械风险管理解决方案需要涵盖所有相关范围。
● 风险分析:
– 危险
– 可预见的事件序列(有时也被定义为根本原因序列)
– 危险情况
– 危害
● 风险评估:
– 缓解前和缓解后发生率值
– 危害严重性
– 风险优先级别
– 判断风险可接受性
● 风险控制:
– 设计要求
– 实现要求
– 标签要求
– 对实施进行验证
– 对有效性进行验证
● 收尾与报告:
– 产品残余风险评估
– 评估风险可接受性
● 上市后监管:
– 风险走势代码
– 风险分析走势代码可追溯性
风险管理系统应该向产品设计师提供以下信息以供采取行动, 包括:
● 危害情况控制计划 – 用于引导危害情况控制计划制定的数据,包括设计、产品实现和标签带来的风险缓解
● 现场性能 – 现场性能必须与风险分析关联起来,从而确保分析过程中考虑这些问题并迅速集成产品使用过程中发现的问题
● 缓解 – 性能数据,包括要求的制定,从而确保验证与确认成果得以运用并且各项要求会有效实施
如果使用电子表格方法追踪风险分析、控制、报告和监管的所有相互关系, 很容易就会不堪重负。使用 Siemens Digital Industries Software 针对医疗器械的解决方案及其关系数据结构、可实施的工作流、自动化、可追溯性与报告,使得风险管理所需数据的追踪和报告变得极为简单。解决方案的中央资源库可以确保每个人使用的数据集相同。只要按下按钮,针对工作项运行报告,系统会填充相关数据,就可以轻松识别危害和其他问题项。
可追溯性是医疗器械风险管理的基础所在。从设计、开发和制 造执行到上市后监管, 企业必须具备精确追踪相关工作项及其与设计和法规要求、测试用例、验证与确认过程、版本控制、危害情况和危害缓解关系的能力。如图 2 所示,Siemens Digital Industries Software 医疗器械模板的综合可追溯性表格中包含一些基本的可追溯性构建块。流程图显示制定设计 验证与确认测试计划和风险管理框架的基本元素。
这种相互关系的复杂性是电子表格或手写文档无法合理追踪的。例如,在设计阶段早期,可以用 Word 创建的文档指定用户需求。由于列出了每一种用户需求,因此在句尾可能需要加一个编号, 以方便追踪。编写特定产品需求文档时,可以重新参考每项用户需求。这样就需要将带编号的链接放置到电子表格中,从而追踪用户需求和产品需求之间的关系。
但是,在设计过程中,您可能无法让所有设计输出,包括子装配都关联到每种用户需求或产品需求。
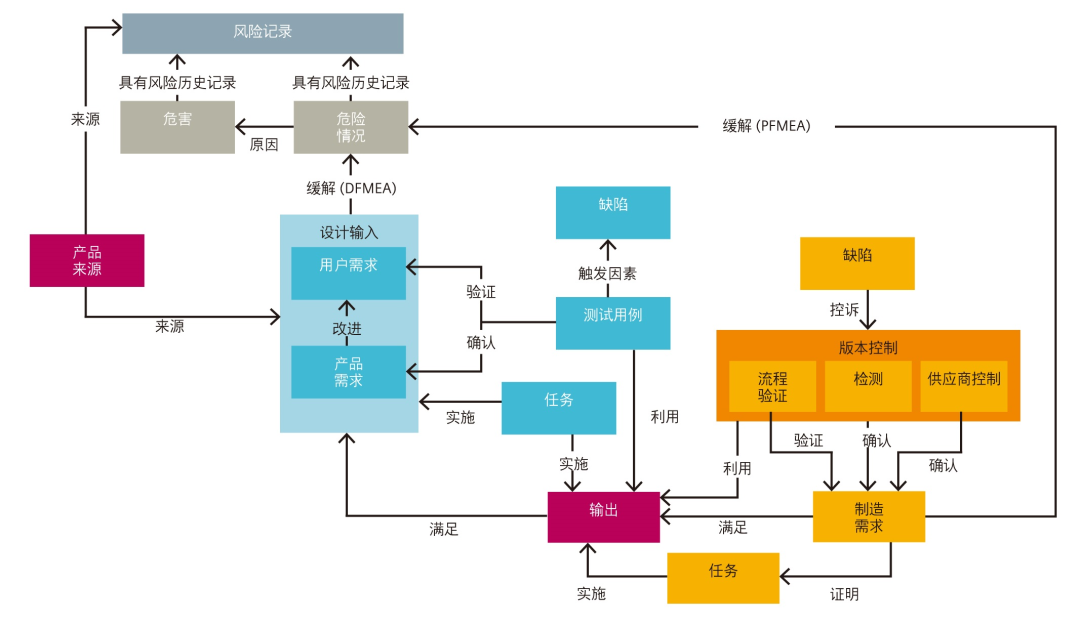
当设计文件输出交接到生产阶段时,追踪变得更加复杂,通常包括创建产品实现计划和电子表格,从而追踪其特定的生产顾虑。因此,为了确保可追溯性符合用户需求、产品需求和制造需求输出,就必须打开所有这些文档。这些文档可能尚未更新、未集中存储或缺少版本控制,因此需要人工查找相关对象并辨认每个文档的可追溯性。
有效可追溯性的缺乏,给尝试响应纠正措施 / 预防措施 (CAPA) 需求的团队带来很多的苦恼和延误。
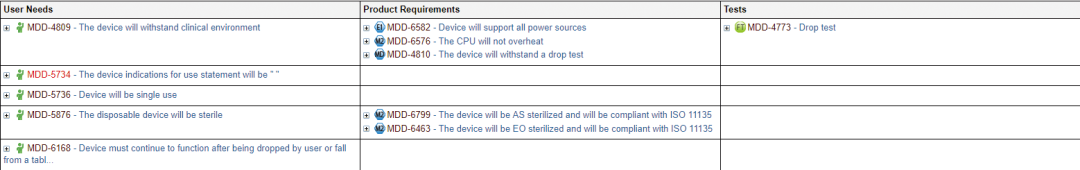
如图 3 所示,高效的产品需求管理解决方案应该便于关联用户需求和产品、软件需求与相关测试。
例如,图 3 表格中的前几个条目显示用户需求 1010-3873(设备必须消过毒)与产品需求 1010-3874(根据 ISO 11135 标准, 设备必须用环氧乙烷灭菌)匹配并引用测试 (1010-3821)。
功能强大的需求管理解决方案支持整个设计过程、医疗器械产品开发过程中典型制造和风险管理关系的工作流和可追溯性。这种解决方案应该支持开发过程中使用的各种概念的融合,例如各种标准的融合、美国食品和药物管理局 (FDA) 的指导方针、ISO、美国材料试验协会 (ASTM) 和所有其他适用合规性数据以及基于图像和文本的证明。
强大的需求管理工具最有效力的一个利用点就是辅助项在整个设计历史记录文件 (DHF) 中的引用方式。
假设以医疗器械预期用途声明为例。工具应该支持已经批准的定义,这样随时可以在使用过程中引用已作标签的工作项。这可以确保文本的一致性以及确定使用标准文本位置的能力。这对于确定变更的全部影响至关重要,也可以确保变更可以恰当填充到所有相关文档。
强大的追踪和追溯功能对于记录和缓解危害情况而言必不可少, 能够控制结果并确保设备具有可预测性和安全性。
我们已经知道,有效而高效的任务管理系统的实施,特别是基于传统电子表格和静态文档的系统实施,纷繁复杂而富有挑战性。
● 根据数据审核方式对其进行组织:将产品发放到现场之后,通过上市后监管根据用户危害以及现场发生率的数量或百分比评估产品。数据字段应该与返回的数据直接关联,以便于轻松比较和响应现场识别到的问题。数据字段应该能够看到危害发生率并可以直接与风险管理过程进行比较。这样可以迅速评估用于确定危险情况影响危害频率的因素是否正确,或者发生危险情况的可能性是否得以正确判断。
● 使用数据字段的公共术语:监管机构已定义危害或危险情况含义。应该将规定的术语融合到模型中,这样系统才能帮助审计人员更好地理解设计意图而无需额外解释。
● 使关联复杂性降至最低:工作项的组织方式应该能够使关联复杂性降至最低。系统可能提供很多自由度 (DOF),以致其逻辑性难以理解。这也给针对系统的员工培训以及向监管机构解释流程的工作带来难度。
风险管理方法通常理解不够充分且定义不够明确,这也使得可靠的风险管理模型变得更为重要。
设计精良的综合风险管理工具应该能够支持风险管理数据模型, 从而确保定义、工作项和工作流使用一致,能够识别和缓解危害。
风险管理工具应该支持图 4 所示的逻辑流,这是根据 ISO 14971 附录 E 派生而来的,是美国和欧洲医疗器械审批系统合规性所必需的。此工具囊括例如事件序列、发生危险情况的概率以及危险情况可能导致危害的概率等衡量要素。
风险管理数据模型带来运营精确度。它可以根据正在处理的设计或法规要求设置定义,然后强制使用的一致性,从而极大地改进数据质量。
使用 Siemens Digital Industries Software 医疗器械解决方案创建风险管理数据模型,可以为团队所有成员制定可实施的工作流, 无论他们位于何处或者存在哪些变量,都可以使用已定义的相同工作流。贵公司可以根据需要创建任何工作流。关键在于,只要确定工作流,就可以确保其使用一致。
这样可以提供统一的框架,帮助确保所有任务都可以毫无遗漏地完成,并且随意定义或不完整的任务不会引入其他变型。
在可实施的框架中定义工作流的过程明确了识别潜在危害并确定更佳缓解策略的相关工作。
这种框架也帮助实现了自动化。一旦建立这种框架,就可以创建工作项。可以使用与电子表格所用相同的评估流程识别危险情况, 不同的是,所有相互关系都是受管的。这可以大幅减少执行计算、追踪和其他往往较为耗时的任务所需的时间。在未来的类似产品中重新使用这些需求和关系,还可以极大地减少工作量。
必须强调的一点就是,电子表格并非框架。它只是单元格的集合, 无法用于实施工作流或处理关系数据。
西门子解决方案所提供的框架及其支持环境还让您可以确保其他元素的一致性,包括定义、术语和拼写。尽管强制执行一致的拼写和术语貌似微不足道的,但如果缺乏一致性就会削弱数据功能。
如果在数据库中搜索肠穿孔之类危险情况,假如 “穿孔” 一词打错了, 只能在 30% 的电子表格条目中看到一部分这样的意外事故。风险管理工具可以强制实施一致的拼写,这样搜索就能识别所有事故。
出于同样的原因,还需要强制实施一致的术语。例如,对于动脉球囊损伤,有些事故可能将其描述为球囊移位,而其他人可能将同样的事故定义为球囊扩张,因此可能产生一系列不同的变化。从一开始就统一使用适当的术语,框架就可以强制使用一致的术语,同时为适应和正确保存事故变型提供灵活性。
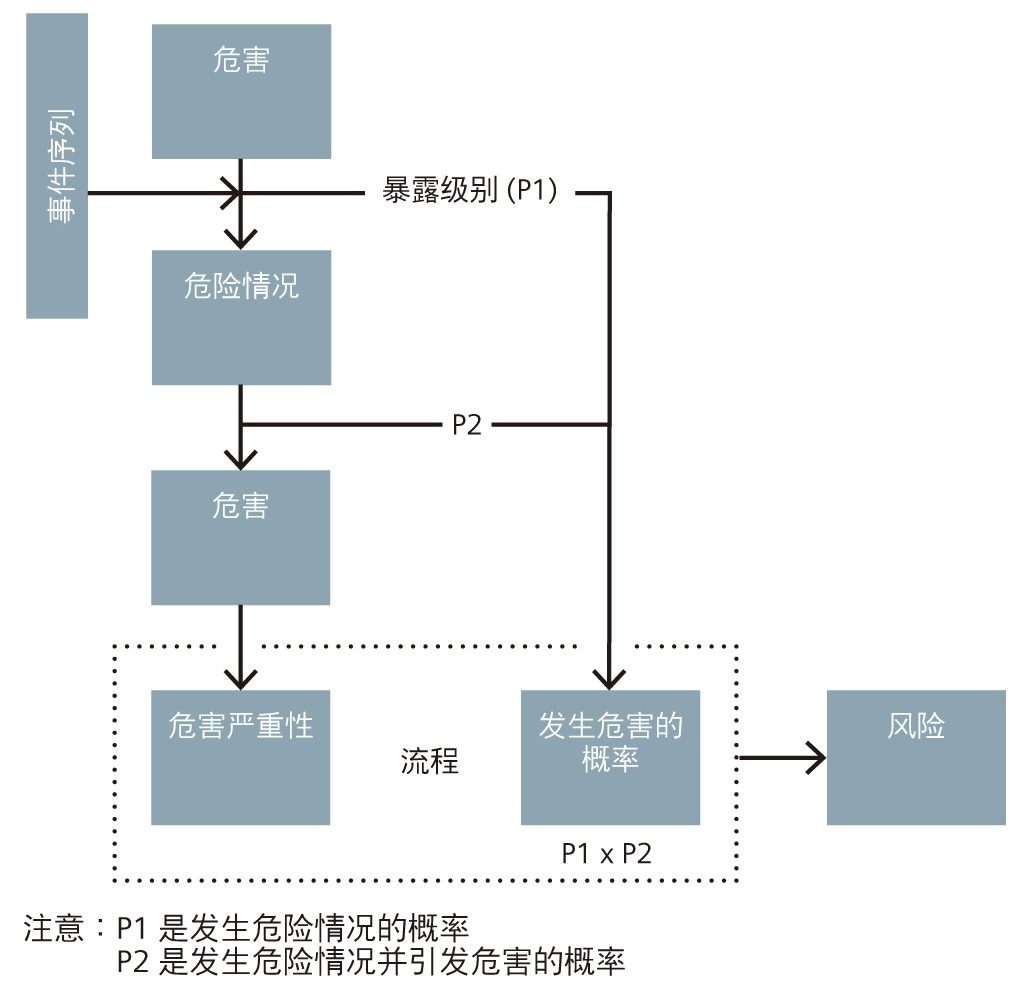
图 4. 基于 ISO 14971 附录 E 的风险管理逻辑流
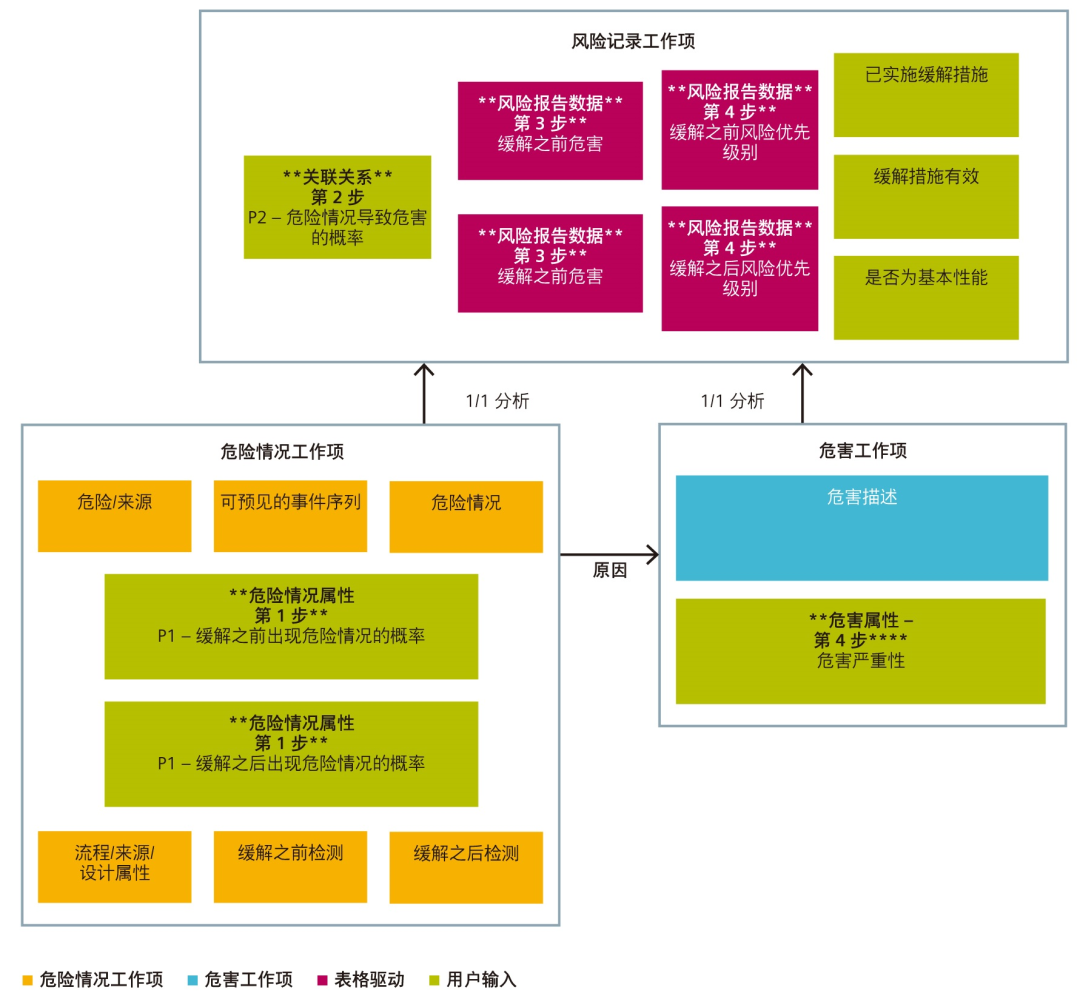
实施系统的风险管理逻辑流要求定义多种工作项和相称的变型, 从而有逻辑地组织各种分析。图 5 提供了符合之前图 4 所示风险管理流程图的示例。该系统根据三个工作项进行组织:风险记录、危害与危险情况。
整体系统分析以传统故障模式和影响分析 (FMEA) 形式进行。
● 工作由不同部门完成和审核,每个部门都使用自己适当的工作流。这种分歧是逻辑性的,因为危险情况主要是工程行为,而风险分析一般由风险管理专业人员和临床医师进行。
● 如果没有工作项规定的组件,则无法定义转变 / 概率变量;例如, 如果不知道危害、可预见的事件序列和产生的危险情况,就无法定义发生危险情况的概率。在风险分析中,如果不收集危险情况发生率和危害特性,就无法确定可能导致伤害病人的危险情况。
● 例如,危险情况术语在法规文档 (ISO 14971) 中有所定义, 因而方便将工作项与法规术语匹配起来,便于进行审计解释工作。
可以定义多种工作项类型以满足一系列需求。正如之前提到的, 三个基本要素包括危害工作项、危险情况工作项和风险记录工作项。
风险评估工作项包括危害描述与危害严重性。例如,过敏性休克的危害描述可能指派给从 1 到 5 的严重性值中的 4。
特性完整的危险情况包括产生故障模式(危害)的来源、故障模式描述(可预见的事件序列)以及局部效应(危险情况)。可变输入包括缓解之前和缓解之后发生危险情况的概率 (P1) 以及缓解之前和缓解之后检测。
● 电磁辐射 > 1) 切口绝缘,2) 导体接触事例 > 底盘柜起电或
● 生物相容性、过敏性 > 1) 注射器端孔超出规范、太大;2) 施用剂量过多 > 病人使用剂量过多
风险记录工作项将一种危险情况与危害结合起来,便于成对比较。在此阶段要完成多个操作,才能完成风险评估。
P2 因素被定义为危险情况与危害之间的关系(危险情况导致危害的概率)。在上述示例中,危险情况是底盘起电。危害是对用户产生的电击。显然,问题在于,用户发生电击导致死亡的概率如何?幸运的是,人们懂得吸取别人的教训。我们可以使用此P2 转换因素将发生率降低到用户能够接受的水平。
P1 和 P2 因素随后可以结合起来,用于确定发生危害的情况。
最终的 P 因素随后会与危害严重性一起使用以确定危害 / 危险情况风险指数。
风险优先级别或风险指数也会被计算,用以确定产品和公司系统中风险可能带来的影响。
定义风险管理系统之后,还需要分级评定。以下内容介绍了每个分级及其含义。这些分级只对行为方式给出示例。
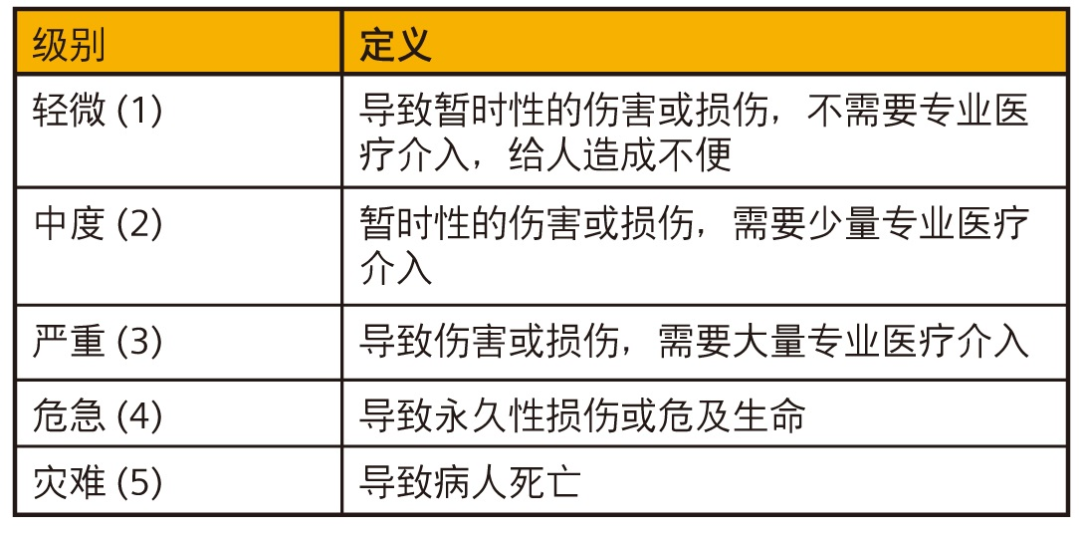
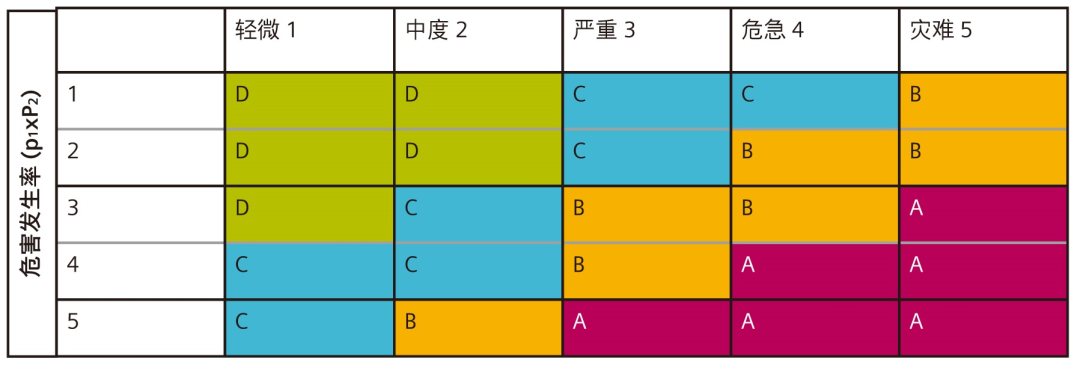
在图 6 规定的系统中,危害严重性按从 1 到 5 这五个级别定义。
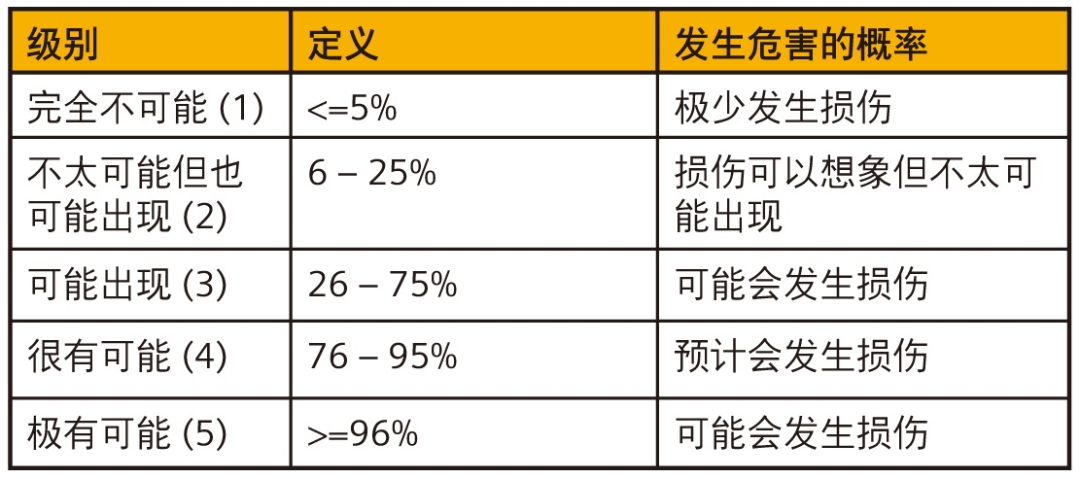
危险情况发生率根据概率划分级别。图 7 所示表格提供此类评级系统示例。
危险情况造成危害的概率也按可能性评级。图 8 显示了一个相关示例。
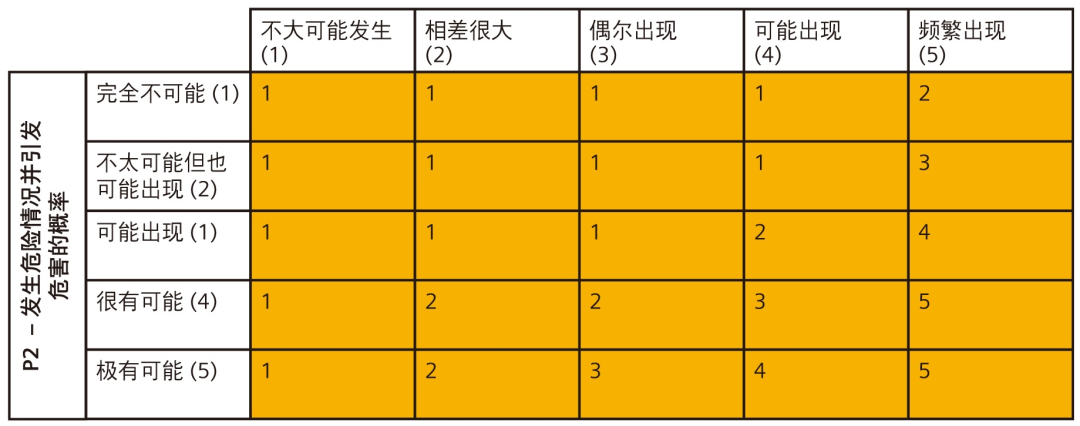
如图 9 所示,通过确定以上定义的 P1 和 P2 发生率值,可以预估发生危害的概率。

风险优先级别 (RPL) 可根据上表确定的严重性和发生率级别计算。可以派生自选取表格或者是多种计算方法。选取表格定义如图 10 所示。
一旦确定每种危险 / 危害组合的特性,就可以生成风险管理和缓解评估报告。此类报告的部分屏幕截图如图 11 所示。
失效模式及影响分析 (FMEA) – 危害/危险和缓解评估
在医疗器械风险管理整个过程中,定义都是必不可少的,包括定义所需文档的结构和内容,例如风险管理计划、风险分析和风险管理报告。
有些文档结构是法律要求的。当然,必须格外小心,确保这些文档包含法规所需的所有信息。
正如 EN ISO 14971:2009 定义的,风险管理计划应包括:
ISO 要求风险分析文档采用基于危害情况的方法。风险分析要素包括:
● 基于危害情况故障树分析 (FTA):数据库可追踪性表格
正如 EN ISO 14971:2009 定义的,风险管理报告应该能够帮助确保:
使用 Siemens Digital Industries Software 解决方案, 可以将美国联邦公报信息提取为需求文档工具,并将它们与公司标准作业程序 (SOP) 要求关联起来。将两者关联起来,可以帮助确保满足公司 SOP 要求或 ISO 标准所需的法律要求,这反过来会为公司设计文档 SOP 提供合规性支持。
此过程确定从需求来源到设计文档证据的审计可追踪性。在审计过程中,公司可以直接从法规要求追踪 SOP,证明公司对于美国联邦法规 (CFR) 或欧洲医疗设备管理局 (MDD) 某个特定章节的合规性,从而便于识别能够证明合规性的记录。
法律要求→
公司 SOP 要求/ISO 标准 →
所有项目文档记录 →
产品实现数据 →
上市后监管数据
风险管理文档应该为明确定义的元素提供恰当的集成,包括:
● 危害危害应该位于综合产品风险分析的首位。除了法规这样要求以外,它也为上市后审计追踪带来便利。如果现场发生不良事件,这些事件通常都与给用户造成的危害有关。在这些情况下,由于风险管理文件是根据危害分类的,可以迅速确定哪些危险情况预计会造成目前的危害,也可以查看为控制危害所采用的所有缓解措施。这为识别是否已考虑问题的根本原因以及纠正现场问题所需措施,提供了一种简便的方式。它还可以迅速识别与设计特征相关的设计验证与确认测试以及在需要进行设计更改的事件中需要重复哪些测试。
● 危险情况:需要从所有可能方面分析危险和危害情况,从而决定设计、制造和设备使用中的潜在问题。所有这些方法(DFMEA、PFMEA、UseFMEA、故障树、临床应用评估、临床试验及其他) 都可用于识别危险情况,使其在基于危害的分析中可被视作用户危害的潜在原因。
● 缓解措施:公司对危险情况处理采取的每一项缓解措施都应该列在基于危害的故障树分析中。用户需求、产品需求和制造需求都应视作危险情况的合理缓解措施。部分原因在于,ISO 14971:2012 附录 Z 要求标签不应降低危害发生率。无法通过设备设计控制使用时,我们需要尽可能全面的策略来控制产品使用。缓解危险情况时,我们需要使用公司可用的所用工具来降低风险, 按照 ISO 14971:2012 的说法是,“ 尽可能地” 降低风险。
● 产品标签:风险缓解策略还是产品标签更好的数据来源。如果并非使用目前市面上售卖的类似器械或是邀请众多医生来定义有关注意事项、警告或禁忌症这些风险,除了风险分析以外, 还有哪些更好的方式来制定全面的潜在问题列表呢?确定高 / 中度风险之后,其中一项缓解措施就是使用产品标签。尽管标签的使用并不会降低危害发生率,但从产品责任角度而言,这无疑是证明何时、何处需要使用用户通知的绝佳方式。
● 设计输入:文档集应该至少包含一个文档,更可能包含多个文档,用于定义用户需求和产品需求,正如产品需求文档 (PRD) 中定义的一样。这些文档通常制定用于反映开发流程以及开发流程中的供应商。此外,还要考虑文档在项目承包环境中的组织方式。
● 设计输出(规范):规范形式多样,包括印刷品、代码与制造工作说明。应该制定有关追踪所有设计要求是否均已满足的计划。根据测试策略的不同,时常并不需要将所有规范都纳入设计控制,并不需要涉及所有文件。另一方面,如果所有规范都纳入系统,就可以追踪产品质量计划(首件检验、在制试验、到货检验)所需所有数据的测试,从而提供整个设备生命周期更为完整的情况。使用这一策略,可以让制造团队将上市后测试数据融合到产品历史记录。
● 设计验证和确认计划:正如前面提到的,搜索用户需求、产品需求和测试用例项目文件的功能是一项强大的功能。
● 产品实现计划 – 该计划用于定义如何构造产品。公司一般会将此列表分解为多个文档。
● 产品构造流程图 – 流程图为后续有关产品制造每个步骤的流程和需求提供了讨论背景。随后将检查此列表是否存在重复, 使其成为主验证计划的基础。需要进行流程验证时,就要定义测试用例。如果不需要进行流程验证,文件需包含不验证的原因说明。
● 主(流程)验证计划 – 此部分文档为方便讨论产品构造使用的所有流程、识别需要验证的所有流程以及放置流程验证测试工作项容器提供了场所。流程验证工作是对潜在产品危害的一种缓解措施,应与基于危害的故障树分析中的危险情况关联显示。
● 质量管理计划 – 一旦确定构造进程,就可以使用质量管理计划,通过构造计划中的产品性能验证要点,为产品质量提供保障。这些验证要点缓解了潜在的产品危害,应该列在基于危害的故障树分析中。
● 设计传递计划 – 一旦产品开发与测试完成并获得批准,对于获批构造供对外使用的产品,设计就必须传递给制造部门。
此计划还谨慎考虑了企业应该如何监控和收集设备制造和现场性能数据。
每次质量管理审核会议中,都应对公司质量方针目标予以评估。这些目标包括质量审计性能、CAPA、客户投诉和制造系统。理想 情况下,设计缓解策略应该设置框架来确定风险更大的领域并提供控制的检查要点。
如果这些测试包含在设计控制框架中,测试数据自然会填充到管理审核流程中。应该对危险情况发生率制定表格,使得现场危害 / 危险情况的审核变得简单而直观。
可视报告工具为项目经理确定每项报告所需的格式和数据提供了极大的灵活性。这种灵活性也为更改不符合预期的结果或复杂结果的报告和数据结构赋予强大的功能。报告输出和背景数据填充需仔细分析,在大量数据集上实施工具之前需对更改进行测试。
● RPL 计算 – 计算产品风险优先级别的唯一方法。计算 RPL 可以使用多种方法。严重性 X 发生率,严重性 2 X 发生率,严重性 X 检出率 X 发生率,这样就会生成多种多样的评级值:1 到10、1 到 5、1 到 20、选取列表、方程和其他变量。实际上, 实现这种功能的方式很多,您可能希望创建自己的定制 RPL 以满足公司特定需求。
● 关联关系 – 有些人构建系统是从产品需求向上追溯到危害, 有些人是从危害向下追溯到缓解措施。如果内部系统是固定的, 则可能需要重新构建关系,使其符合贵公司的 SOP。好消息是, Siemens Digital Industries Software 解 决 方 案 灵 活, 支 持任一种方法。
● 工作项术语 – 相同逻辑概念时常可能使用不同的术语。让系统顺应公司方针就显得理所当然。
● DFMEA、PFMEA 术语 – FMEA 已经存在一段时间,但其工具的使用在不同行业变化多端,有时不同术语的使用也会渗透到医疗器械行业。如何将 FMEA 呈现并发布到设计控制文件,也是需要考虑的事项。
● 设计可追踪性报告 – 设计可追踪性报告是对设计打样的描述。如果任意构建组件(工作项、关联关系、背景数据)发生变更, 则报告和呈现的关联格式也需要更改。工作项审批工作流正是良好的例证。
毫无疑问,成功完成医疗器械开发项目所需的系统架构极易让项目经理应接不暇。起初,项目经理可能会说,“这有何难?我做 个表格就行了”,随后就开始使用 Excel? 电子表格软件进行设计输入,并使用 Word 开始传输产品需求文档。但是,只要稍加研究开发流程的复杂性,就会发现,这一过程会造成工作量的不断增加,几何数量的增加还会导致出错概率提高。
Siemens Digital Industries Software 解决方案可以在管理复杂设计工件和关联各种关系方面,助您一臂之力。采用此解决方案,已经确立的各种关系会保存无损而无需耗费大量维护精力,同时项目更新仍然可以在结构化、可搜索的环境中进行。







 苏公网安备 32059002002276号
苏公网安备 32059002002276号