



背景:气体在有机溶剂、聚合物或沸石中的扩散率可以通过运行分子动力学模拟并确定气体在材料中的均方位移来计算。这允许您计算气体的自扩散系数,并深入了解整体扩散率。在进行分子动力学计算时,可以分析温度、压力、密度以及渗透剂的尺寸和结构对扩散的影响。
简介:在本教程中,您将通过构建一个包含甲烷和PBD的无定形单元来计算甲烷在聚(顺式-1,4-丁二烯)(PBD)中的扩散率。在您构建完单元后,您将执行分子动力学模拟,并计算甲烷分子的均方位移。虽然本教程将只演示一个简短的计算,但您将会熟悉所涉及的方法。或者,您可以通过Pipeline Pilot运行整个工作流。该教程基于Meunier (2005年)发表的一篇论文,该论文研究了气体在二烯聚合物中的扩散。
目的:介绍使用力场方法计算气体在致密材料中的扩散系数。
本教程重要节点:
建立最初的结构-建造一个无定形的晶胞-弛豫晶胞-运行和分析分子动力学-在Pipeline Pilot中运行整个工作流
1. 建立最初的结构
第一步是构建和优化甲烷分子和PBD聚合物的结构,以便构建无定形晶胞。为了便于之后选择甲烷分子,可使用不同的显示样式。
使用均聚物Homopolymer构建工具从dienes库创建20个重复单元的c_丁二烯聚合物c_butadiene。
创建一个新的3D原子文档3D Atomistic Document并绘制一个甲烷分子。将原子的显示样式Display style更改为CPK。重命名新文档。
提示:不要从示例结构库导入甲烷。该示例没有将结构定义为文档层次结构中的分子,这将在以后尝试选择所有甲烷分子时出现问题。
在本教程中,将在静电和范德华相互作用的计算中使用电荷组。电荷组是净电荷为零的较小原子片段。这意味着两个组之间的静电相互作用可以用一个简单的基于距离的截断值来评估,避免了计算上更昂贵的Ewald方法。
如果需要,可以手动设置电荷组,但自动分配的电荷组通常可以达到较高的计算要求。首先,将Forcite参数设置为在非键计算中使用电荷组而不是默认的求和方法。
在菜单栏中选择Modules | Forcite | Calculation,打开Forcite Calculation对话框。在Energy选项卡中,从Forcefield下拉列表中选择COMPASSIII。将Electrostatic和van der Waals求和方法均设置为Group based。
单击Forcefield后面的More…按钮,打开Forcite Preparation Options对话框。确确保Charges设置为Forcefield assigned,并对Forcefield types和Charge groups均勾选了Calculate automatically复选框。关闭对话框。
在建立无定形晶胞之前,应对分子进行几何优化。
在Forcite Calculation对话框的Setup选项卡中,从Task的下拉列表中选择Geometry Optimization。将Max. iterations更改为2000。
现在优化甲烷分子。
使得methane.xsd为当前文档,单击Forcite Calculation对话框中的Run按钮。
在Project Explorer将建立一个名为methane Forcite GeomOpt的新文件夹。当计算结束后,优化好的结构将保存在该文件夹中。继续优化聚合物结构。
使得Polyc_butadiene.xsd为当前文档,单击Run按钮。关闭Forcite Calculation对话框。
将重复相同的过程,优化后的结构将返回到Polyc_butadiene Forcite GeomOptPolyc_butadiene.xsd文件。
现在可以观察由计算自动分配的电荷组。
使得Polyc_butadiene Forcite GeomOptPolyc_butadiene.xsd为当前文档。在3D Viewer中空白区域右键单击,从弹出的快捷菜单中选择Display Style,打开Display Style对话框。将Color by选项更改为Charge Group。确认电荷组分配后,将Color by选项更改回Element。对methane Forcite GeomOptmethane.xsd重复此过程,并关闭对话框。
注意:聚合物中的扭转自由度将由Amorphous Cell模块修改。将在构建晶胞后进行优化。
在继续进行建立无定形晶胞之前,清理工作区。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
2. 建造一个无定形的晶胞
当两个模型已经构建完成后,即可使用Amorphous Cell模块在晶胞中构建它们的多个副本。
单击Modules工具条上的Amorphous Cell按钮,在下拉列表中选择Calculation。
打开Amorphous Cell Calculation对话框。
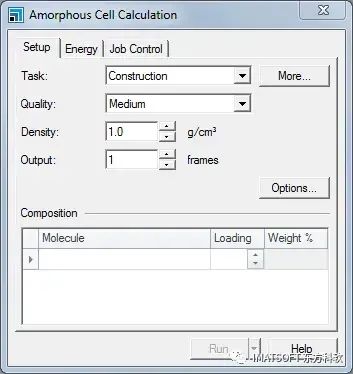
Amorphous Cell Calculation对话框的Setup选项卡
第一步是根据每种组分的分子数量来确定成分。晶胞中需含有4个甲烷分子和10个PBD分子,密度为0.95 g/cm3。
将Density设置为0.95 g/cm3。
在组分Composition列表的Molecule列中,选择包含甲烷优化后结构的文档methane Forcite GeomOptmethane.xsd。在Loading列中输入4。
在下一行中,选择Polyc_butadiene Forcite GeomOptPolyc_butadiene.xsd,加载量为10。
根据每个分子的加载量和晶胞的密度,晶胞的估计尺寸显示在对话框底部。在这种情况下,将构造单元长度约为27 Å的立方体。也可使用正交晶格和四方晶格类型,但本教程中未使用。
在建立无定形晶胞时,也可用Amorphous Cell模块优化结构。本例中,将使用Forcite单独优化和平衡构型,而不使用此功能。
单击Options…按钮,打开Amorphous Cell Options对话框。取消勾选Optimize geometry复选框。关闭对话框。
现在选择与Forcite计算中相同的力场。
在Amorphous Cell Calculation对话框的Energy选项卡中,从Forcefield下拉列表中选择COMPASSIII。
Amorphous Cell中的默认计算任务描述为第一个组分的名称,在本例中为methane,在所有输出文档中用作seedname。在本教程中,将默认设置更改为cell。
在Job Control选项卡中,取消勾选Automatic复选框,在文本区域输入cell。单击Project Explorer中gas_polymer树形图根目录,单击Run按钮。关闭对话框。
在Project Explorer将建立并显示一个名为cell AC Construct的新文件夹。当计算结束后,将产生一个包含无定形晶胞的轨迹文件cell.xtd。
注意:如果构造了多帧,它们都将存储在.xtd文档中。可以使用Animation工具栏查看和访问。
双击cell.xtd。
该文件包含一个含有10个PBD低聚物和4个甲烷分子的周期性晶胞。
在以下步骤中,使用结构文档(.xsd)而不是轨迹文档(.xtd)中的模型更为方便。所以应该在一个新的3D原子文档中复制该结构。
右键单击轨迹文件空白区域,选择Copy选项进行复制。
从菜单栏中选择File | New…,选择3D Atomistic文档,单击OK按钮。右键单击新文档中的空白区域,选择Paste粘贴已经复制的结构。将新文档重命名为cell。
在继续进行晶胞弛豫之前,清理工作区。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
3. 弛豫晶胞
当建立一个无定形晶胞时,分子可能不会均匀分布在整个晶胞中,从而形成低密度的区域。要调整这一点,必须执行较短时间的能量最小化(几何优化)以优化晶胞。在能量最小化之后,应该运行一个较短的分子动力学模拟来平衡晶胞结构。这种最小化和分子动力学的过程被称为结构弛豫,应该在构建无定形晶胞时进行。
要执行几何优化,必须首先将使用三维周期结构的电荷组配置Forcite参数。
重新打开新创建的结构文档cell.xsd。打开Forcite Calculation对话框并选择Energy选项卡。将Electrostatic和van der Waals求和方法更改为Group Based。
注意:Forcite对非周期结构和周期结构有单独的设置。该对话框始终显示与活动文档的周期性相对应的设置,默认为非周期性。
现在,已经准备好最小化晶胞的总能量。
在Forcite Calculation对话框中,单击Run按钮。
计算任务完成后,最终结构将存储在cell Forcite GeomOpt文件夹中。将通过在周期性变化的温度下运行分子动力学来继续弛豫这个结构,也就是对体系退火。在本教程中,将只运行一个退火循环。
在Forcite Calculation对话框中的Setup选项卡中,从Task下拉列表中选择Anneal,单击More…按钮,打开Forcite Anneal Dynamics对话框。
将Annealing cycles的数值设置为1,Initial temperature设置为300 K,Mid-cycle temperature设置为500 K。关闭对话框。
现在,在优化后的结构上运行退火计算。
使得cell Forcite GeomOpt子文件夹中的cell.xsd为当前文档。单击Forcite Calculation对话框中的Run按钮。
退火计算任务会产生几个输出文件。最后一次温度变化后的最终结构包含在cell Forcite Anneal文件夹下的cell.xsd文件中。将继续在这个结构上运行一个恒温下较短时间的分子动力学模拟。
使得cell Forcite Anneal子文件夹中的cell.xsd为当前文档。在Forcite Calculation对话框的Setup选项卡中,从Task下拉列表中选择Dynamics,单击More…按钮,打开Forcite Dynamics对话框。
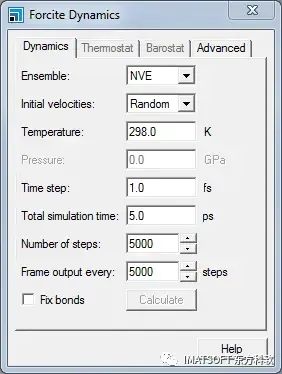
Forcite Dynamics对话框的Dynamics选项卡
有不同类型的分子动力学模拟可用,按系综名称可分为NVE、NVT、NPT和NPH。其中字母表示:
N=恒定分子数
V=恒定体积
E=恒定能量
T=恒定温度
P=恒定压强
H=恒定焓值
注意:如果建立模型时选择的密度(即体积)需要调整到与外部压强一致(通常为大气压强),则应使用NPT系综;如果体系以合理的密度构建,则可以使用NVT,且平均压强应为1 atm或0.0001 GPa。
已经以0.95 g/cm3的密度构建了晶胞。由于这也是该体系在300 K和1 atm下的平均密度,在选定的力场下,无需使用NPT进一步弛豫密度。
因此,可以使用NVT系综运行分子动力学模拟。
从Ensemble下拉列表中选择NVT,并将Temperature更改为300。
在本教程中,将把运行步数减少到2000。对于这种较短时间的分子动力学运行,速度标度Velocity Scale恒温方法比默认值更合适。
将Number of steps更改为2000。在Thermostat选项卡上,选择Velocity Scale作为恒温方法。单击Forcite Calculation对话框上的Run按钮。
注意:在真实模拟中,可能需要运行至少50000次运行步数(50 ps)才能正确平衡晶胞。可以通过在实时更新的图表中查找能量来研究平衡过程,除了小的波动,该图表应该是恒定的。
模拟完成后,将返回大量文档。最终结构包含在cell Forcite Dynamics文件夹中的结构文档cell.xsd中。清理工作区。
从菜单栏中选择File | Save Project,然后选择Window | Close All。
4. 运行和分析分子动力学
当平衡体系结构时,只对最终的构型感兴趣。但是,要计算晶胞中甲烷分子的均方位移,需要有许多帧构型,以便可以分析甲烷分子移动的位置。将运行另一个分子动力学模拟并生成轨迹文档,可以使用Forcite分析工具对其进行分析。
为避免子文件夹过多,可首先将工作文档移动到文件夹树的顶部。
在Project Explorer中,双击cell Forcite Dynamics文件夹中的cell.xsd文件。在Project Explorer中,将此文件拖动到gas_polymer文件夹树的顶部。
之前,已经在恒温(NVT)下运行了动力学计算,但是对于产生构型的计算运行,将在恒定能量(NVE)下继续模拟。这是因为某些恒温方法可能会干扰系统的动力学,并可能影响稍后将计算的扩散系数。为了收集足够的数据进行分析,应增加计算步数并缩短每帧输出间隔。在本教程中,只需执行5000步。
在Forcite Dynamics对话框的Dynamics选项卡上,从Ensemble下拉列表中选择NVE。将Number of steps更改为5000,Frame output every更改为250。关闭Forcite Dynamics对话框。
在Forcite Calculation对话框中,单击Run按钮并关闭对话框。
提示:对于实际产生构型的计算运行,应该增加步骤数,使得模拟时间至少为50 ps。
随着计算的进行,将更新两个图表文档。一个绘制总能量和各种成分随时间的变化,另一个绘制温度随时间的变化。因为这是一个NVE系综计算,所以总能量应该是恒定的。会有动能和势能的交换,但如果平衡时间足够长,就不会有净交换。同样,在平衡体系中,温度将在平均值300 K左右波动,无系统变化。
计算完成后,将返回包含21帧的轨迹文件cell.xtd,并对其执行分析。
使得cell.xtd成为当前文档。单击Animation工具栏上的Play按钮。观看完轨迹动画后,单击Stop按钮。
要计算甲烷分子的均方位移,需要将它们与聚合物分子区分开来。这可以通过将它们定义为一个集合来实现。
要选择所有甲烷分子,请按住CTRL键并依次双击每个分子。
提示:要自动选择一种类型的所有分子,可以使用Find Patterns工具。
现在可以使用Edit Sets工具创建一个选定的原子集合。
从菜单栏中选择Edit | Edit Sets以打开Edit Sets对话框。单击New…按钮,输入名称methane,然后单击OK按钮。关闭Edit Sets对话框。单击轨迹文档中的任意位置以取消选择。
既然已经将甲烷分子定义为一个集合,就可以分析它们的运动了。
单击Modules工具条上的Forcite按钮,在下拉列表中选择Analysis。打开Forcite Analysis对话框。
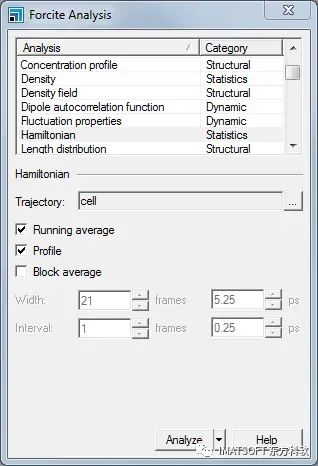
Forcite Analysis对话框
可以用Forcite进行许多不同类型的分析,它们被分为三类;结构Structural、统计Statistics和动力学Dynamic。均方位移位于动力学Dynamic部分。
从列表中选择Mean square displacement。从Sets下拉列表中选择methane,将Length设置为21。
单击Analyze按钮,关闭对话框。
注意:由于MSD数据的统计准确性随着时间间隔的长度而降低,因此默认情况下,MSD的计算所使用的帧数不会超过总帧数的一半。
Forcite Analysis工具计算均方位移并生成图表文档cell Forcite MSD.xcd,其中包含甲烷分子随时间的均方位移(MSD)图。以及数据表文件cell Forcite MSD.std。图表中报告的给定时间的MSD值是该长度的所有时间间隔和集合中所有原子的平均值。
均方位移通常有两个区域。在短时间内,气体分子在一个自由体积的小区域里碰撞。由于分子受到限制,它不会在这个时间尺度上扩散,MSD水平保持恒定。在更长的时间尺度上,分子从受限区域跳到另一个自由体积的区域。由此产生的重复跳跃运动是扩散,其特征是均方位移在时间上是线性的。在实践中,统计数据会随着时间间隔而减少,通常会在结束时产生较大的波动。
MSD随时间的增加与扩散系数D有关:
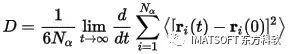
其中Nα是体系中扩散原子的数量。生成MSD数据时,Forcite中的MSD会自动计算该数量,并在图表文档Forcite MSD.xcd标题中列出扩散系数,并将其输出在数据表Forcite MSD.std的Summary表单中。
扩散系数以Å2/ps为单位计算。要转换为更常用的单位cm2/s,结果值必须除以1e4。
甲烷在PBD中扩散的计算值在2.25×10-6 cm2 s-1和7.5×10-6 cm2 s-1范围(Meunier, 2005)。这是通过4个甲烷分子在一个包含10条由30个重复单元的PBD聚合物链的无定形晶胞的计算中获得的。采用温度循环退火法对晶胞进行平衡。在400至250 K(温度每步变化25 K)的加热和冷却循环下,以5-10 ps的模拟时间内进行了几个NPT动力学循环。接着在250至400 K的模拟温度下,以25 K的增量进行了3 ns的平衡NVT动力学。通过均方位移分析,得到了扩散系数随温度的变化关系。
本例的计算值可能与文献中的值有较大差异,因为运行长度非常短,扩散区域的统计数据有限。在实际中,应该运行更长时间的模拟,并对从独立生成的结构开始的多个分子动力学模拟进行平均,以估计计算的精度。Amorphous Cell模块可以一次生成多个独立的帧,这可能有助于产生模拟结构的研究。
5. 在Pipeline Pilot中运行整个工作流
质量传输特性的计算,如扩散,在材料科学的许多领域中都有广泛的应用。为了简化研究(如本文所述),BIOVIA Pipeline Pilot中的Materials Studio Collection包括一个协议,该协议自动从输入结构列表计算扩散系数。本教程的此可选部分解释如何使用此协议。
创建一个新的数据表文档Study Table Document,并将其命名为gas_polymer.std。
在Project Explorer中定位至文件methane Forcite GeomOptmethane.xsd。在文件名上单击鼠标右键,然后选择Insert Into。对Polyc_butadiene Forcite GeomOptPolyc_butadiene.xsd重复此步骤。
右键单击gas_polymer.std中的B列,选择Properties,然后输入名称loading。在此列的前两行中输入值4和10。
这些步骤为Pipeline Pilot中的质量和电荷传输协议准备了输入文档,将在下一步载入它。
从主菜单中选择Tools | Pipeline Pilot Protocols,并选择合适的Server location。从Protocols | BIOVIA Materials Studio | Battery文件夹中打开Mass and Charge Transport协议。
该协议从创建无定形晶胞晶格开始,正如在本教程前面所做的那样。随后,使用NPT系综进行初始分子动力学运行,以建立收敛的密度、温度和初始速度集合。最后,进行采样以计算扩散系数和相关的传输特性。
下一步是在协议对话框中输入所需的物理参数。
将Temperature更改为300,Configurations更改为4,Target Density更改为0.95。
在Initialization部分,将Time更改为2。
在Sampling部分,将Time更改为10,Frame Interval更改为0.05,Min Rsq更改为0.9。
如果计划在排队系统上运行,请在Run via Grid部分选择适当的选项。
单击Run。
此作业运行四组独立的计算并将结果进行平均。结果以两个PDF报告和包含轨迹数据的轨迹文件夹的形式返回。gas_polymer Initialization Report.pdf文件描述了输入结构的化学性质,并提供了每个单独轨迹初始化的温度和密度图。从这些数据中可见,在本例中,初始化时间太短,无法给出收敛结果。如上所述,进行高精度计算将需要更长的运行时间。
报告gas_polymer Sampling Report.pdf返回所有计算的聚合和平均传输特性,以及每个单独轨迹的分析。报告的初始部分包含适用于整个体系的数据,如密度和温度。还有一个表,其中列出了每个分子的数据,包括扩散系数及其相对浓度。总体汇总包含从样本空间平均中获得的标准误差。对于每个单独的轨迹分析,包括MSD图、相应的直线拟合和拟合的R2值,以帮助评估模拟的质量。
注意:可能会查看到一些标有红色的线,这些线对应于至少一个的分子,其MSD数据拟合后返回的R2值低于最初指定的目标值。为了获得更好的统计信息,可以在启用Extend Trajectories选项的情况下重新启动计算任务。
如果数据表包含带电分子,该协议将计算模拟晶胞的电导率,以及每种成分的部分电导率。此功能设计可用于诸如电池的电解质溶液的分析。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号