



背景:力场是经典模拟计算的核心,因为它体现了结构中每种类型的原子如何与其周围的原子相互作用。对于体系中的每个原子,分配一个力场类型,描述原子的局部环境。力场包含描述各种属性的信息,例如平衡键长度和力场类型对之间的静电相互作用。通常,力场是通用的,以覆盖许多体系,或针对特定问题进行参数化。一般的力场,如Dreiding,在不同环境中有许多原子的参数。但是,对于特定情况,它永远不会像仅针对该情况参数化的力场那样表现良好。因此,能够编辑力场以用于尚未对其进行参数化的体系会很有用。
这可能在许多情况下发生:您可能希望参数化力场以模拟沸石框架中分子的吸附等温线,您可能具有要用于提高多晶型预测计算准确性的实验晶体数据,或者您可能希望更改力场以更好地描述聚合物的密度。在所有这些情况下,您都需要编辑基础力场并观察这些更改的效果。
编辑力场是一项复杂的任务,因为许多交互都是显式或隐式耦合的。例如,旋转扭转角的能量曲线不仅取决于扭转项,还取决于非键能。因此,获取正确的参数可能需要相当长的时间,并且涉及许多不同的计算。最好的方法是进行小的体系性更改,并查看对体系的影响。与所有形式的参数化一样,需要在获得可靠结果和过度拟合消耗资源之间权衡。
简介:在本教程中,您将修改小分子的单个扭转角,以改善优化模型和晶体结构之间的匹配。最初,您将使用Dreiding力场优化爆炸材料三氨基三硝基苯(TATB)的晶体结构。然后,您将修改Dreiding力场以改善晶体结构和力场优化模型结构之间的匹配。您将结合使用Conformers模块来探测扭转能关系,DMol3来提供从头算数据,Forcite来优化晶体结构。
目的:说明了在Dreiding中编辑力场参数的工作流程,以改进计算晶体结构数据与实验晶体结构数据之间的拟合。
本教程重要节点:
优化晶体结构-检查扭转角度-使用非键项-修正范德华项
1. 优化晶体结构
打开Import Document对话框。导航至ExamplesDocuments3D Model文件夹,双击TATNBZ.xsd文件。将结构重命名为TATNBZ_crystal.xsd。
在将修改后的力场应用于晶体结构以验证这些更改之前,将对孤立的分子执行所有力场编辑工作。应该复制一个孤立的分子。
在Project Explorer中,右键单击project目录,从弹出的快捷菜单中选择New | 3D Atomistic。
在TATNBZ_crystal.xsd,单击其中一个分子中的一个原子,右键并选择Select Fragment。按下CTRL + C键。在新建的原子文档中按下CTRL + V键。在Project Explorer中,将新的原子文档重命名为TATNBZ_molecule.xsd。
如果检查晶体结构,会发现硝基在苯环所在的平面上。现在,将使用Forcite模块利用标准Dreiding力场优化结构。
确保TATNBZ_crystal.xsd文件为当前活动文档。单击Modules工具条上的Forcite按钮,在下拉列表中选择Calculation,打开Forcite Calculation对话框。
将Task修改为Geometry Optimization。在Energy选项卡中,将Forcefield设置为Dreiding,并将Charges更改为Charge using Gasteiger。
可以使用更复杂的电荷计算方法,例如使用DMol3应用ESP电荷,但Gasteiger电荷将适用于自定义力场的开发。
单击Run按钮。计算任务完成后,打开优化后的TATNBZ_crystal.xsd文档。
应该看到硝基不再和苯环在处于同一平面上。优化后的结构中硝基的离面性质会影响这些分子在晶体中聚集的方式,因此获得平面硝基非常重要。
硝基和苯环之间有一个单一的扭矩,这将控制基团的平面度。修改控制此扭矩的参数将是本教程的重点内容。
2. 检查扭转角度
现在已经确定了要更改的扭转角,可以开始更详细地查看该扭转角的能量分布。通过计算扭矩旋转360°时的单点能,Conformers计算将提供该能量分布。
右键单击TATNBZ_molecule.xsd,从弹出的快捷菜单中选择Label,打开Label对话框。确保Object type已设置为Atom,从Properties列表中选择Name。单击Apply按钮。
使用Measure/Change工具在硝基上定义原子序列为O5-N5-C5-C4的扭矩。
在Label对话框中,单击Remove按钮,关闭对话框。
将使用Conformers模块系统性地旋转扭矩,并在每点计算能量。
单击Modules工具条上的Conformers按钮,在下拉列表中选择Calculation,打开Conformers Calculation对话框。单击Torsions…按钮,打开Conformers Torsions对话框。
将# Steps修改为72,关闭对话框。
在Energy选项卡中,将Forcefield设置为Dreiding,并将Charges更改为Charge using Gasteiger。单击Run按钮,关闭对话框。
当计算结束后,即可以查看扭矩-能量图,从而观察能量分布。
在数据表中选择B列和C列,单击Quick Plot按钮 。
。
将图表文件重命名为Dreiding_Original.xcd。
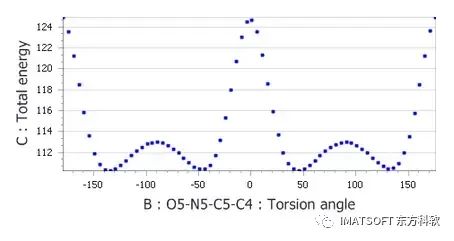
Dreiding力场计算硝基-苯扭转角的扭矩-能量图
从扭矩-能量图中可以看出,在0°和180°处存在两个绝对值较大的能量最大值,这为旋转提供了约14 kcal mol-1的势垒。通过检查晶体结构,可以预期在0°和180°处存在能量最小值,而不是扭矩-能量图所示的最大值。
也可以将这个势能面与DMol3预测的势能面进行比较。由于已经有一个包含构象的数据表,可以使用DMol3模块来计算每个构象的能量。
注意:由于此计算需要精确的能量,将使用较高的精度设置,此计算可能需要一个多小时。ExamplesStudyTables文件夹中提供了包含计算结果的数据表。如果希望使用此文件,请跳过接下来的两个步骤。
在数据表中,选择A列。单击Models按钮 ,
,
打开Models对话框。定位至DMol3 Molecular Energy模型,双击并编辑。
对于该计算,需要精确能量。应使用高精度泛函和收敛标准。
在Model Editor – DMol3 Molecular对话框的Inputs选项卡中,将Functional修改为BLYP,Quality level修改为Fine。单击Save按钮,关闭对话框。在Models对话框中单击Run按钮。
高精度的精度等级不仅更改SCF收敛标准,同时也改变其他设置,如基组。该计算需要一定时间才能完成,建议远程运行或隔夜运行。必须等待计算完成,才能进入下一步,也可导入示例数据表。
打开数据表,或从ExamplesStudyTablesTATNBZ_conformers.std导入示例数据表。
将观察到Total Energy (DMol3 Molecular)的单位为Hartree,因此需将其转换为kcal mol-1 (1 Hartree = 627.51 kcal mol-1)。假设总能量列为列D,则需要定义一个函数D×627.51。一旦完成单位转换,应该像绘制原始构型图一样绘制能量图。
单击Define Function按钮 ,
,
打开Define Function对话框。在Expression文本区输入D*627.51。输入DMol3 Total Energy (kcal/mol)作为Name,单击OK按钮。选择包含扭转的列和新创建的列,单击Quick Plot按钮。
注意:能量的绝对值将非常高,因为它们没有使用DMol3进行优化。然而,这里只关心相对势垒高度,而不关注能量绝对值。
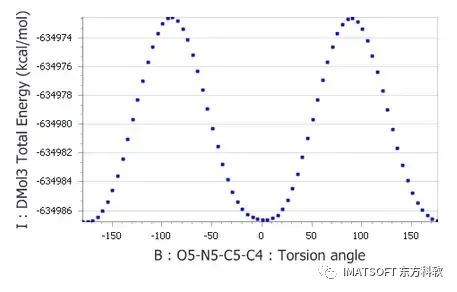
DMol3单点能的二面角-能量图
从图表中可以看出,势垒高度约为14 kcal mol-1。在本教程的后面部分,将尝试从力场计算中获得相同的近似势垒高度。
现在已计算了由Conformers和DMol3预测的能量分布,可以将其与Dreiding中的扭矩项进行比较。Forcefield Manager用于创建标准力场的可编辑副本。
单击Modules工具条上的Forcite按钮,在下拉列表中选择Forcefield Manager。在Standard Forcefields部分,选择Dreiding。单击>>按钮。关闭对话框。
将在工程中创建Dreiding力场的副本,并打开Dreiding.off力场供编辑。力场文档由四个选项卡组成:
Summary – 包含描述力场的摘要文本。
Types – 包含力场类型及其关联的属性。
Interactions – 包含主要的相互作用,如价键项和非键项。
Equivalences – 包含等效相互作用的定义。
编辑力场之前,可以更改力场文档的名称。
在Project Explorer中,将力场文档的名称更改为Dreiding_new.off。
现在已准备好查看力场文件。
在Dreiding_new.off文件中,选择Types选项卡。
其中包含Dreiding中可用的力场类型,以及每个力场类型的类型、元素和范德华参数的说明。可以使用Forcefield Type Properties对话框上的复选框显示其他属性,例如杂化、电荷和氢键。
有许多力场类型描述每个元素的不同局部环境,有些元素具有多个力场类型。滚动浏览所有这些内容会很枯燥,因此可以按力场类型过滤显示的内容。过滤器框在力场文档中显示为黄色。
在黄色的Type Filter文本框中,输入C_*并单击ENTER键。
现在,Types对话框中仅显示与碳原子有关的原子类型。
将Type Filter更改回*。
也可以按3D模型文档中存在的力场类型进行过滤。
勾选Filter by selection in,在下拉列表中选择TATNBZ_molecule.xsd。
将看到一个警告对话框。这是因为分子中的原子没有力场类型。可以使用Forcite Calculation对话框分配力场类型。
打开Forcite Calculation对话框。在Energy选项卡中,单击与Forcefield关联的More…按钮,打开Forcite Preparation Options对话框。取消选择Forcefield types的Calculate automatically复选框,将TATNBZ_molecule.xsd更改为当前文档。单击Calculate按钮。再次勾选Calculate automatically。关闭所有Forcite对话框。
这计算了TATNBZ_molecule中原子的力场类型。需要刷新力场文档中的力场类型。
将Dreiding_new.off更改为当前文档。重新选择TATNBZ_molecule.xsd。
此时可以观察到代表文档中的原子的四个力场类型,和一个虚拟力场类型。
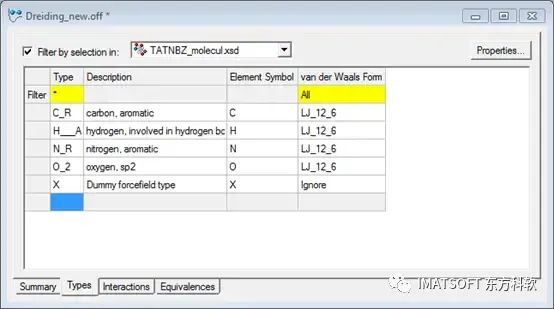
Dreiding_new力场文件,显示了力场类型信息
本例中所关注的是芳香族碳C_R和芳香族氮N_R之间的扭转角。
打开Interactions选项卡。将Show interaction更改为Torsion。
扭矩项有六种不同的组合。可以更改Functional Form过滤器以查看不同的选项。
单击Functional Form过滤器并选择Dihedral。
显示描述扭矩的二面体函数形式中使用的参数值。在线帮助中记录了所有参数。
选择显示的行之一,然后按下F1键。
将显示帮助,显示不同的函数形式。二面体扭转的函数形式为:
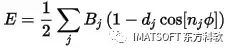
可以使用之前生成的Conformers数据表绘制纯扭矩项的变化,并比较Conformers和DMol3得出的整个能量表达式的函数形式。对于力场相互作用,将上述方程中的Bj替换为25,d替换为1,nj替换为2。将使用数据表中B列的角度值,转换为弧度。
使得TATNBZ_conformers.std为当前文档,打开Define Function对话框。在Expression文本区,输入(25*(1-1*cos(2*B*0.0174532925)))/2。将Name设置为Dihedral from Dreiding,单击OK按钮。选择B和J列,单击Quick Plot按钮。
生成的图表应与下面的图表相匹配,显示与DMol3图相对应的最小值和最大值,但能垒高于预期。
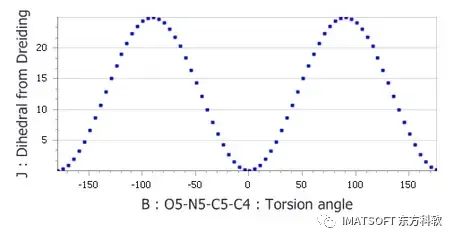
使用X C_R N_R X扭矩力场编辑器得到的纯二面角分项绘制二面角-能量图
从上面的图中,可以看出二面角扭转看起来是合理的。这表明问题一定出在其他地方。在下一节中,将研究非键项,以了解它们如何影响扭转能量。
3. 使用非键项
从前面的计算中,观察到纯二面角的能量贡献与观察到的扭矩旋转时的能量变化显著不同。可以使用力场编辑器关闭体系中不同类型相互作用的贡献,以查看它们的效果。因为只改变了一个末端扭转角,除了扭矩本身的能量,改变的贡献是非键能。因此,将关闭非键能,以查看它们对扭矩-能量图的影响。
由于硝基的氧原子很可能与相邻的胺形成强氢键,因此首先禁用氢键项是有意义的。
打开Dreiding_new.off文档。在Interactions选项卡中,单击More…按钮。
打开Forcefield Preferences对话框,在其中可以设置力场的的一般首选项。
在相互作用列表中取消勾选Hydrogen Bond复选框。
提示:在将力场文档与Conformers或Forcite等模块一起使用之前,无需保存该文档。如果进行了大量更改,建议将其保存。
现在你需要用新的力场运行另一个Conformers计算,以观察关闭氢键相互作用的效果。
在工程根目录中打开TATNBZ_molecule.xsd文件。打开Conformers Calculation对话框,选择Energy选项卡。从Forcefield下拉列表中选择Browse…,打开Choose Forcefield对话框。选择Dreiding_new.off,并确保Charges设置为Charge using Gasteiger。
在Forcefield文本框中将显示Dreiding_new。反斜杠表示已从工程中选择了力场,它不是标准的力场。
注意:此力场将在运行计算时传输到服务器,并将返回到新创建的计算结果文件夹中。
在Conformers Calculation对话框中,单击Run按钮。
当计算任务结束后,打开TATNBZ_molecule.std文档,对B和C列绘制曲线图。将其与Dreiding_Original.xcd进行比较。
应注意到禁用氢键相互作用对扭矩-能量图的影响微乎其微。接下来,试着金庸静电相互作用。
将Dreiding_new.off文档改为当前文档。在Forcefield Preferences对话框的相互作用列表中取消勾选Electrostatic复选框。将工程根目录中的TATNBZ_molecule.xsd更改为当前文档。打开Conformers Calculation对话框,选择Energy选项卡。
修改过的Dreiding版本仍然为所选力场,因此可以仅运行计算任务。
运行Run Conformers计算。当计算任务结束后,绘制扭矩和能量关系的曲线图,将其与Dreiding_Original.xcd进行比较。
可见,与打开静电相互作用相比,能量较低的极大值在高度上有所降低。另外,总能量增加了几kcal mol-1。
最后,关闭范德华相互作用以查看效果。
将Dreiding_new.off文档改为当前文档。在Forcefield Preferences对话框的相互作用列表中取消勾选van der Waals复选框。对TATNBZ_molecule.xsd文档运行Conformers计算。在输出数据表中,绘制扭矩和能量关系的曲线图,将其与Dreiding_Original.xcd进行比较。
当关闭范德华相互作用项时,应在扭矩-能量图上看到重大的变化。能量分布应该与通过二面角相互作用生成的曲线图非常相似。
这表明范德华项在这个扭转角的能量分布中起着主要作用。修改范德华项将是下一节的重点。
将Dreiding_new.off文档改为当前文档。在相互作用列表中勾选van der Waals、Hydrogen Bond和Electrostatic复选框,关闭Forcefield Preferences对话框。
去除范德华势后产生的变化与硝基上的氧原子和胺基上的氢原子之间的近距离有关。通过修改范德华项,使其具有较短的平衡距离,从而在较短的距离内减少排斥力,应该看到,Conformers得到的图开始与DMol3预测的更接近。
在工程根目录中将TATNBZ_molecular.xsd设置为当前文档。使用Measure/Change工具在氧原子和相邻氢原子之间添加Distance测量。
距离应约为1.8 Å。在下一节中修改范德华项时,将使用此信息。
4. 修正范德华项
Dreiding中的范德华项是根据每种力场类型的参数组合自动生成的。由于参数存储在每个力场类型中,因此它们将显示在Types选项卡上的力场文档中。还可以为原子对添加自定义范德华项,这些项将被用来代替自动计算的项。
在Dreiding_new.off文件中,选择Types选项卡。单击van der Waals Form的黄色过滤器框,然后选择LJ_12_6。
注意:可以选择显示函数形式,也可以Ignore相互作用。如果选择Ignore,则不会在能量表达式中计算相互作用。为van der Waals力场类型选择Ignore,将忽略包含该类型的所有范德华相互作用。尽管这在本例中看起来过于激进,但如果想关闭单独的相互作用,ignore可能很有用。
可以设置函数形式,也可以选择忽略范德华参数。Dreiding使用简单的Lennard Jones 12-6形式计算范德华参数,其中D0为阱深,R0为平衡距离。
记下H___A和O_2的D0和R0值。
可见与O_2相比,H___A的阱深非常浅。参数组合通常使用算术平均值计算。然而,由于氢的阱深非常小,在这种情况下,阱深参数的几何组合可以更好地表示范德华相互作用。
几何平均值计算为阱深乘积的平方根,对于H___A和O_2等于0.0031。
打开Interactions选项卡。将Show interaction更改为van der Waals。将Functional Form更改为LJ_12_6。
注意到,没有为Dreiding定义特定的范德华项。要引入特定的范德华项,必须首先指定力场类型序列。
在Fi选框中单击第一个空行,然后从下拉列表中选择H___A。单击Fj选框并从下拉列表中选择O_2。在Functional Form选框中单击并选择LJ_12_6。将D0的值设置为0.0031。
对于该测试,应该使用算术平均值计算R0。该值为3.29。
将R0的值设置为3.29。在工程根目录中的TATNBZ_molecule.xsd文件中运行Conformers计算。生成扭矩-能量图。
可见极小值的高度已经改变了约0.5 kcal mol-1,但对曲线图没有太大的总体影响。改变函数形式的另一种方法是减小平衡距离的值。之前,计算了分子中氧原子到氢原子的距离。这约为1.8 Å,而R0为3.29 Å。这表明范德华势的排斥项对扭矩势有重要贡献。要纠正此问题,应减小R0的值。虽然很容易将平衡值设置为测量距离的平衡值,但这可能会过度拟合相互作用,从而导致整体结构的变化。事实上,选择正确的数字可能需要一些猜测,但最好先较高值开始逐渐降低,运行多次Conformers计算。在本教程中,使用了2.7 Å的R0值。
在Dreiding_new.off中,将R0的值更改为2.7。在TATNBZ_molecule.xsd中运行Conformers计算。生成扭矩-能量图。
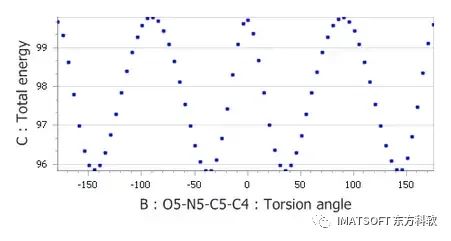
力场编辑器中调整X C_R N_R X扭矩的范德华参数后的二面角-能量图
曲线图显示,改变平衡位置对扭转角的能量分布有显著影响,在0度和180度时能量最大值显著降低。然而,总体能量势垒现在明显低于先前DMol3计算中预测的能量势垒。为了补偿这一点,应该增加扭转能量势垒。
在Dreiding_new.off中,将Show Interaction更改为Torsion,Functional Form过滤器更改为Dihedral。将B的X N_R C_R X值更改为50。在TATNBZ_molecule.xsd中,运行Conformers计算。产生扭矩-能量图。
可见,新的扭矩-能量图现在与从头算计算预测的结果相似,扭转势垒的大小和位置也相似。在0度和180度的两侧仍有两个低能极小值,这将导致结构变形。然而,改变H___A O_2范德华相互作用的值将不再有太大的影响。
这表明小的排斥项一定是由另一个原子与硝基上的氧相互作用引起的。
将工程根目录中的TATNBZ_molecule.xsd文件打开为当前文档。在氧原子和最近邻胺基氮原子上添加一个Distance测量。
距离应约为2.5 Å。与氢-氧相互作用一样,这小于力场中的平衡距离。该距离意味着范德瓦尔斯斥力将很大,这可能导致势能面上的绝对值较小的最大值。
将Dreiding_new.off打开为当前文档。选择Types选项卡,记下N_R和O_2的D0和R0的值。
与氢和氧范德华项一样,将使用几何平均值计算新的D0项,并将R0设置为2.7 Å。
打开Interactions选项卡。仅显示van der Waals相互作用。单击黄色过滤器选框Functional Form,选择LJ_12_6。单击空白的Fi选框,选择N_R。单击空白的Fj选框,选择O_2。在Functional Form选框中单击并选择LJ_12_6。将D0的值设置为0.086,R0的值设置为2.7。在TATNBZ_molecule.xsd中,运行Conformers计算。产生扭矩-能量图。
不应存在额外的最小值,图表的形状应与之前生成的DMol3图相似,最小值略为平坦。然而,当改变氧和氮的范德华参数的函数形式时,二面角的能量势垒现在太高了。根据DMol3计算,希望其约为14 kcal mol-1。
将Dreiding_new.off文档改为当前文档。在Interactions选项卡中,选择Torsion相互作用。在Functional Form中,选择Dihedral。将B的X N_R C_R X值更改为35。在TATNBZ_molecule.xsd中,运行Conformers计算。产生扭矩-能量图。
这将得到约为14 kcal mol-1的势垒。可以继续调整参数以进一步改善力场,但现在的力场应该足以测试晶体结构。
打开原始的TATNBZ_crystal.xsd文件。打开Forcite Calculation对话框。选择Energy选项卡,选择Dreiding_new.off作为力场。单击Run按钮。
当计算完成后,可见硝基几乎在苯环平面上排列,就像它们在晶体结构中一样。
从这里,可以使用新的力场运行多晶型预测Polymorph Prediction计算。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号