


背景:Materials Studio 中的 Forcite 是一款经典的分子力学工具,由 BIOVIA 科学家和软件工程师设计,用于执行一系列任务,包括单分子和周期系统的快速能量计算和几何优化。它提供对不同力场的访问,并且对于新手来说很容易使用,并且为有经验的用户提供最广泛的自定义选项。
简介:对表面相互作用和反应的详细了解在许多材料和工艺的设计中起着关键作用。第一步是准备一个物理吸附到表面的分子模型,该模型具有优化的几何形状(即能量最小化)。建模方法涉及不同步骤,包括从纯晶体构建表面、在表面附近添加分子、定义势(通过力场)来研究气固相互作用,然后是几何优化计算。本教程旨在解释研究气体在表面上的物理吸附所涉及的不同步骤。本教程中的体系是一个受钨表面吸附H2案例研究启发的示例(参见 White (1996) 论文)。在这种特殊情况下,分子力学的使用可以被视为计算成本更高的量子力学方法的先驱:一旦使用 Forcite 优化模型,就可以使用 DMol3 或 CASTEP 来研究化学吸附反应。
目的:介绍使用 Forcite 在表面上生成优化的分子结构
本教程重要节点:
开始-从纯晶体构建金属表面-将气相分子置于表面-执行 Forcite几何优化
1.开始
首先启动 Materials Studio 并创建一个新项目。
打开 New Project 对话框并输入 H2_W 作为项目名称,单击 OK 按钮。
新项目是使用 Project Explorer 中列出的 H2_W 创建的。下一步是加载要切割的晶体结构,在本教程中,您将使用纯钨 (W)。
单击Import按钮以打开“导入文档”对话框。导航到 Structures/metals/pure-metals 文件夹并双击 W.msi。
钨的晶体结构显示在名为 W.xsd 的 3D 原子文档中。
2.从纯晶体构建金属表面
从菜单栏中选择Build | Surfaces | Cleave Surface。
这将打开 Cleave Surface 对话框。
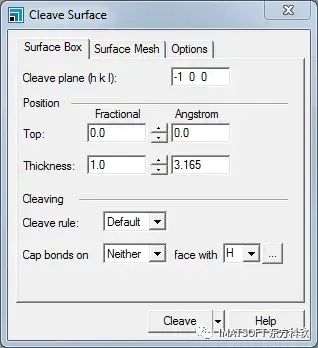
“Cleave Surface”对话框,“Surface Box”选项卡
蓝色虚线显示在晶胞上,表示您正在切割的平面。这些信息也在Surface Box选项卡的顶部给出。默认平面为-1 0 0,相当于(1 0 0)曲面,因此不需要更改此设置。
您应该改变晶胞的厚度,以确保最终模型包括气体分子和钨原子之间所有重要的相互作用。
增加Fractional Thickness到2.0。
厚度应该是6.33 Å。既然已经设置了表面参数,就可以切割晶胞了。
单击Cleave按钮并关闭对话框。
打开一个新的3D Atomistic文档W (-1 0 0).xsd,一个2D周期结构显示为一个带有钨原子的白色正方形。
现在您已经用一个3D晶体构建了一个2D单元。然而,在将气体分子放置到表面之前,需要增加表面的横向范围。这样做是为了避免优化过程中气体分子与周期性图像之间的相互作用。
从菜单栏中选择Build | symmetric | Supercell。
这将打开Supercell对话框。
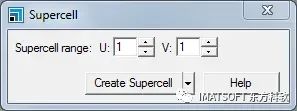
超晶胞对话框
将U和V的Supercell范围增加到4。点击Create Supercell按钮并关闭对话框。
建立了一个由16个原始单元组成的较大曲面,该模型具有二维周期性。您将使用Build Vacuum Slab功能将其转换为3D点阵。
平板是一种三维周期晶胞,其表面在底部,上面是真空。这使得您可以把分子放在真空的表面上,这样分子就不会接触上面表面的周期性图像。
从菜单栏选择Build | Crystals | Build Vacuum Slab…。
这将打开构建真空板晶体对话框。
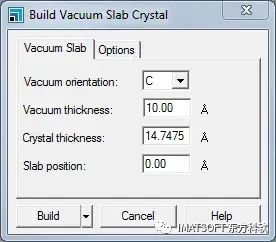
建立真空板晶体对话框,真空板选项卡
这个对话框允许您指定真空区域的方向、厚度和位置。
将Slab position从0.00更改为1.00 Å。增加Vacuum thickness到20.00 Å。单击Build按钮。
W (-1 0 0) 3D原子文档更新显示一个矩形框,其表面位于底部。钨原子在晶胞内,使它们更容易形象化。
3.将气相分子置于表面
本教程的下一个阶段是建立一个氢分子,然后把它放在表面上。要做到这一点,您将在一个新的3D原子文档中描绘一个氢分子,复制并粘贴到平板上,并停靠在表面上。
创建一个新的3D原子文档和草图,并clean一个H2分子。
下一步是将其复制并粘贴到包含slab的文档中。
从菜单栏中选择Edit | Copy。在项目资源管理器中双击W (-1 0 0).xsd,然后选择Edit | Paste。
提示:您也可以使用标准的Windows快捷键CTRL + C进行复制,CTRL + V进行粘贴。
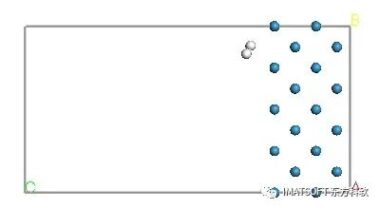
氢分子被粘在晶胞里。由于周期性边界条件,第二个”ghost”分子也出现了。以后可以很容易地删除这个周期性图像。
您可以使用Close Contacts工具来监测氢气离表面有多近。
从菜单栏中选择Build | Close Contacts以打开密切联系计算对话框。选中Monitor close contacts复选框并关闭对话框。
现在您必须选择氢分子平移和旋转它直到它与金属表面对接。
选择hydrogen。按住SHIFT和ALT键以及鼠标右键。移动鼠标来翻译分子。
如果您有一个三键鼠标或带滚轮的鼠标,您可以使用滚轮或带SHIFT键的中间键来翻译,而不用按住ALT键。
提示:当您移动氢原子时,一个粉色虚线就会出现,表示一个密切接触。使用这些控件来定位氢分子刚好在钨表面的上方,粉色线刚好显示出来。
您可以通过更改显示样式来删除相邻单元格中的”ghost”分子。
在3D查看器中单击右键,从快捷菜单中选择Display Style,打开显示样式对话框。在Lattice选项卡上,从Style下拉列表中选择“In-Cell”。在Atom选项卡上,选择Ball and stick选项。关闭对话框。
单击3D查看器中的任何位置以取消选择所有内容。
4.执行 Forcite几何优化
本教程的最后一步是计算氢分子在钨表面的实际物理吸附。
使W (-1 0 0).xsd为活动文档。使用3D Viewer工具栏上的Reset View工具将模型放置在窗口的中心。
单击Modules工具栏上的Forcite按钮,选择Calculation或从菜单栏中选择Modules | Forcite | Calculation。
这将打开Forcite计算对话框。
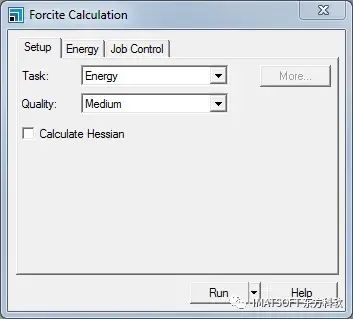
计算对话框,设置选项卡
将Task由能量转向Geometry Optimization,质量Quality to Fine。
点击More…按钮显示Forcite几何优化对话框。将Maximum number of iterations更改为1000并关闭对话框。
默认使用的最小化算法是Smart算法;它是使用最陡下降和共轭梯度算法的层叠方法。
在Energy选项卡上设置Forcefield为Universal。
执行几何优化之前的最后一步是确定金属原子的坐标。通过这样做,您假设表面结构与体积相似,没有明显地被氢原子改变。为了更精确和详细的研究,可能需要在进行几何优化之前放松晶体表面。
按住ALT键,双击任意一个钨原子。所有的钨原子现在都高亮显示。
从菜单栏中选择Modify | Constraints,打开编辑约束对话框对话框。在Atom选项卡上选中Fix Cartesian position复选框并关闭对话框。
单击3D Viewer中的任意位置以取消选择所有原子。
现在可以运行模拟了。
点击Forcite Calculation对话框中的Run按钮。
在项目资源管理器中打开一个名为W (-1 0 0) Forcite GeomOpt的新文件夹。计算时间应在1分钟内完成,并达到规定的收敛准则。完成后,靠近新文件夹顶部的W (-1 0 0).xsd文档包含优化后的结构,应该类似于下面所示的结构。
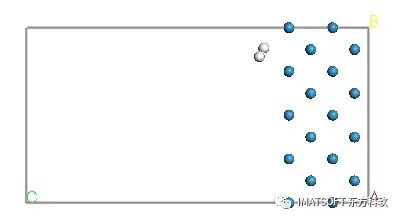
在W (-1 0 0) Forcite GeomOpt文件夹中还有其他四个文档。W (-1 0 0).txt文档包含作业的所有文本信息,特别是初始结构和最终结构的结构和能量参数值。txt文档包含最后一个作业执行状态。W(-1 0 0)能量图表示优化过程中总能量的变化情况。W(-1 0 0)收敛图显示了收敛准则Energy Change和Gradient Norm随优化步骤的变化情况。一旦满足所有要求的标准,模拟就会停止。
现在,您已经准备了一个结构,它将非常适合于进一步的模拟,例如使用DMol3或CASTEP模块的解离反应。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号