




所需功能和模块:Discovery Studio client,DS_Ludi,DS_De novo evolution
所需数据文件:1fvv_protein.dsv, 1fvv_ligand.dsv
所需时间:10分钟
介绍
随着蛋白质结构解析技术的发展以及人类基因组计划的开展,对蛋白质晶体结构的研究发展迅速。怎样有效的分析这些靶点的信息以及设计出相匹配的分子是计算化学家面临的很大挑战。基于分子对接的数据库搜索方法一般是基于已有的数据库,分子结构是不发生变化的。从头药物设计则是一个全新的概念。
Ludi是应用最为广泛的从头设计方法之一。使用Ludi来发现新的具有潜在活性的化合物,可节省研究者大量的时间。Ludi强大的设计工具允许使用者在实验分析之前模拟筛选,并允许对已有的化合物进行改造。Ludi易于操作,它包含有drug-like片段库,同时也允许用户将自己的分子片段加入到片段库中。
DS中的De Novo Protocols使用的即为Ludi的算法,它帮助我们找到新的分子骨架或者修改已有的分子骨架来提高小分子的活性。De Novo可以帮助我们进一步发展已经商业化的分子骨架数据库,并且通过对药物候选分子衍生物的打分来修饰已知的分子配体。这些Protocol包括De Novo Receptor可以用来寻找合适受体结合位点的潜在碎片,这种方法的优点是可以快速有效的对数据库中大量的碎片进行筛选;De Novo Link可以将找到的活性片段连接或在已有骨架的基础上添加新的片段分子;De Novo Evolution也称为AutoLudi,主要用于进行Me better药物设计工作,不仅可以搜索活性片段分子并且可以将片段和骨架自动连接直接产生新的分子。
本教程中,用从头药物设计的方法设计细胞周期蛋白依赖性激酶2(CDK2)抑制剂。
准备蛋白受体和骨架配体分子
运行De Novo Receptor及结果分析
运行De Novo Link及结果分析
运行De Novo Receptor及结果分析
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Lead Optimization,点击 De Novo Receptor工具栏,打开参数面板。
确认Inupt Receptor设置为1fvv_protein:1fvv_protein。
点击Input Fragment Libraries参数,浏览,选择fragment_all.inp。
注意:本例中使用预先产生的片段库,该库可以通过De Novo Library Generation protocol产生。每个片段库包括两个文件,structure file 和target file,同时产生,选其一即可。
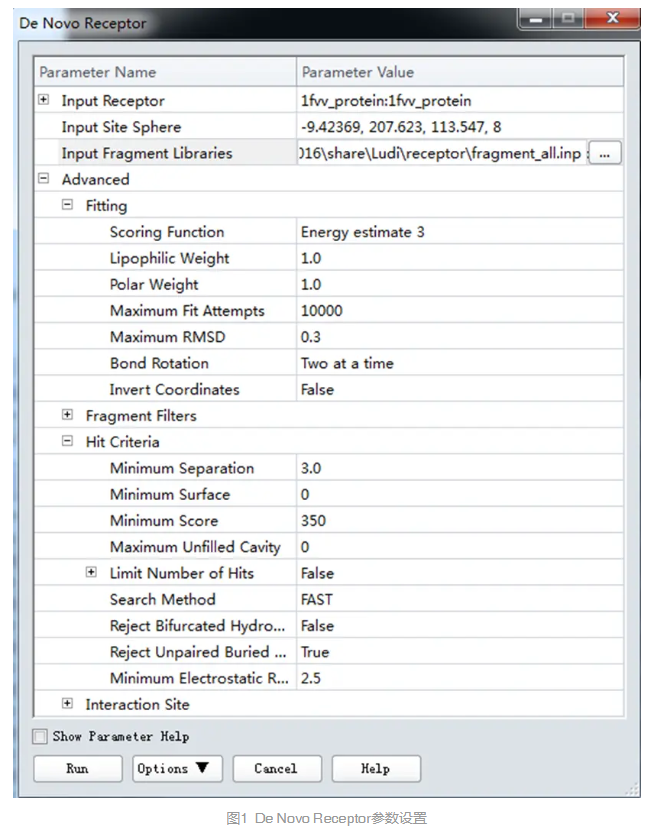
为了减少计算时间,本教程通过修改默认参数来限制hits数。
展开Hit Criteria参数组,设置Minimum Score为350。
展开Fitting参数组,设置Maximum RMSD值为0.3。
其它参数均使用缺省值(图1)。
点击Run按钮,等待计算完毕。
完成该作业大约需要1分钟。
待作业完成后,DS会自动跳出一个新的以1fvv_protein命名的分子窗口。
点击表格视图

和将各片段在受体结合位点中的可结合位置在图形窗口中显示出来。
展开菜单栏Files |Insert From…,点击Files,找到Samples| Tutorials | Receptro-Ligand Interactions | 1fvv_ligand.dsv。
将天然配体显示在同一窗口中。
在表格视图(Data Table )中将1fvv_ligand的Visibility Lock设为Yes。(图2)
这样就会在视图窗口中锁定该天然配体。

在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | View Interactions,点击Ligand Interactions。
受体与配体分子片段间的非键相互作用会以虚线显示出来(图3)。

点击上述View Interaction工具面板下的

按钮可以观察不同的片段在受体结合位点中的可结合位置、同天然配体的叠合情况以及其与受体间的非键作用。
注意:也可在View Interaction工具面板下点击Interaction Options…,来显示指定的非键作用类型。
此外,在表格里包含了各分子片段的Ludi_3_RMSD等碎片信息,以及每个碎片的打分情况。也可以通过在Graphics View中重叠结果文件中的interaction map (输出文件中Ludi.interactionmap.mol2文件)来进一步观察碎片的位置。
双击结果Output文件夹中的Ludi.interactionmap.mol2文件或者点击Jobs Explorer中该任务下的Interaction Map链接,将显示相互作用位点图。
这是分析受体活性口袋内的氨基酸残基的化学特征示意图,其中“蓝白线”代表此位置与受体匹配的相应配体基团应为氢键供体;“灰红线”则代表此位置的配体基团为氢键受体;灰色的点代表配体中的碳原子。(图4)。
注意:如果找不到Output文件夹的路径,则可以在任务浏览器(Jobs Explorer)中,选中相应的任务,然后点击鼠标右键选择Locate即可找到相应Output文件夹的位置。

注意到有些对接片段和天然配体结构叠合的很好,而有些对接片段所在位置则没有天然配体,这就表明受体结合位点并没有被天然配体完全占据,而这也可以被用来优化配体分子以加强同受体的结合。
运行De Novo Link及结果分析
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Fragment Based Design,点击 De Novo Link,打开参数面板。
确认Inupt Receptor设置为1fvv_protein:1fvv_protein。
点击Input Ligand Scaffold参数,选择1fvv_ligand:1fvv_ligand。
点击Input Fragment Libraries参数,浏览,选择fragment_link.inp。
注意:本例中使用预先产生的片段库,该库可以通过De Novo Library Generation protocol产生。每个片段库包括两个文件,structure file 和target file,同时产生,选其一即可。
确保Link Points参数设置为Use all hydrogen atoms。
所有H原子都是片段连接的潜在位置。
为了减少计算时间,本教程通过修改默认参数来限制hits数。
展开Advanced|Fitting参数组,设置Fitting Maximum RMSD值为0.3。
其它参数均使用缺省值(图5)。

点击Run按钮,等待计算完毕。
完成该作业大约需要1分钟。
待作业完成后,DS会自动跳出一个新的以1fvv_protein_complex命名的分子窗口。
点击表格视图中
和将各片段在受体结合位点中的可结合位置在图形窗口中显示出来。
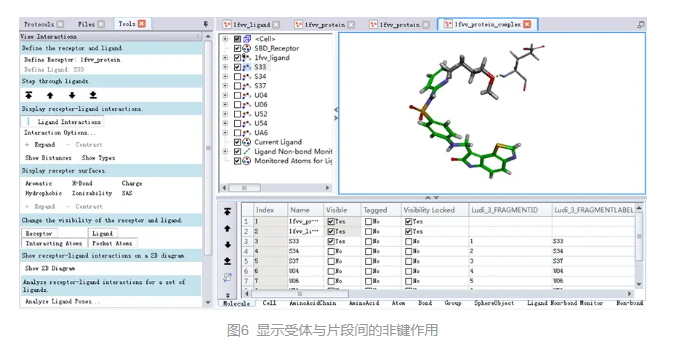
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | View Interactions,点击Ligand Interactions。
受体与配体分子片段间的非键相互作用会以虚线显示出来(图6)。
点击上述View Interaction工具面板下的
按钮可以观察不同的片段在受体结合位点中的可结合位置及其与受体间的非键作用。
双击结果Output文件夹中的Ludi.interactionmap.mol2文件或者点击Jobs Explorer中该任务下的Interaction Map链接,将显示分析化学特征的结果。
将该interaction map拷贝至De Novo Link结果窗口中,可以查看配体分子片段同interaction map的叠合情况,进而分析片段在受体结合位点中的位置。
图15为分析受体活性口袋内的氨基酸残基的化学特征示意图,其中“蓝白线”代表此位置与受体匹配的相应配体基团应为氢键供体;“灰红线”则代表此位置的配体基团为氢键受体;灰色的点代表配体中的碳原子;黄色的双行线代表片段的可连接位置。由于“Link Points”选择了程序默认的“Use all hydrogen atoms”,因此黄色线即为先导分子氢原子位置。

注:片段终端原子连接至骨架分子的H原子处,这也表明可以将片段分子和骨架分子进行共价连接,提高配体分子同受体的结合。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号