




目的:通过此教程,了解Discovery Studio中构建基于受体三维结构药效团的操作方法及结果分析。
所需功能和模块:Discovery Studio client,DS CATALYST SBP,DS CATALYST SCORE,DS CATALYST CONFOMATION。
所需数据文件:1k22_potein_ligand.dsv,thrombin_ligands.sd,kinase_ligands.sd。
所需时间:30分钟
介绍
来自学术界和制药企业的信息表明,Catalyst是更好的基于小分子结构的药物设计工具。然而,科学家的愿望不仅仅是这些,大家也非常希望更大限度地利用已知的受体结构和药物-受体结合信息,有效地克服现有药物设计工具的缺点,更快地加速新药研发的过程。
Structure Based Pharmacophore(SBP)利用已知或假设的蛋白活性位点从受体位点的特性直接得到相互作用位点图,并且将这个信息转化成适用于快速三维数据库检索的药效团模型。这些相互作用位点图还可以进行编辑、分类,以确保只有较重要的信息会被保留下来用于虚拟筛选。同时SBP中还可考虑受体活性位点处氨基酸残基的空间排布,使得通过虚拟筛选得到的分子不仅满足和受体结合位点化学特征上的互补,还可以在空间上很好地结合在活性位点空腔处。SBP引入了那些使用受体结构信息的更佳方法,这些方法已经在高通量筛选的实验环境中所使用。
SBP方法首先通过对活性部位的分析产生特征相互作用图(feature interaction map),特征相互作用图用于反映可能的配体与蛋白之间的合理的相互作用,接着通过特征相互作用图产生药效团模型,进一步利用药效团模型发现潜在的活性化合物。
本教程中使用а-凝血酶作为例子,展示如何利用SBP方法建立药效团模型,开展数据库搜索,以及Discovery Studio如何提供了方便的结果分析工具帮助大家获得更好的结果。
本教程包括以下步骤:
· 构建基于蛋白结构的药效团模型
· 编辑药效团模型
· 配体库筛选
· 筛选结果的分析
Structure-Based Pharmacophore的构建
本教程基于а-凝血酶蛋白结构(PDB号:1K22)来构建药效团模。该蛋白结构从PDB库中下载得到并经Prepare Protein预先准备。
1. 导入蛋白及配体结构
在文件浏览器(Files Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开1k22_protein_ligand.dsv。
а-凝血酶是丝氨酸蛋白酶,是胰凝乳蛋白酶家族中的一员。该蛋白共有三个结合口袋,口袋处残基分别用红色、蓝色和绿色表示。(图1)
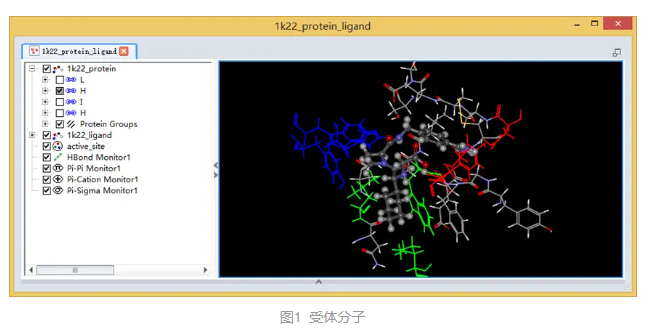 2. 定义活性位点
2. 定义活性位点
在系统视图(Hierarchy View)中选取1k22_protein。
在工具浏览器(Tools Explorer)中,展开Receptor-Ligand Interactions | Define and Edit Binding Site,在工具面板中单击Define Receptor。
将前面选取的蛋白分子1k22_protein定义为受体分子,供下一步使用。
在系统视图(Hierarchy View)中选取1k22_ligand。
在Define and Edit Binding Site工具面板中点击Define Site栏中的From Current Selection。
在视图窗口(Graphic View)中自动生成了一个红色的球,该球将1k22配体包裹其中。同时在系统视图(Hierarchy View)中增加了SBD_Site_Sphere的选项。
如果蛋白结构中没有配体小分子的信息,则可以选择已知活性位点处的残基,或者用binding site工具自动搜索结合位点。
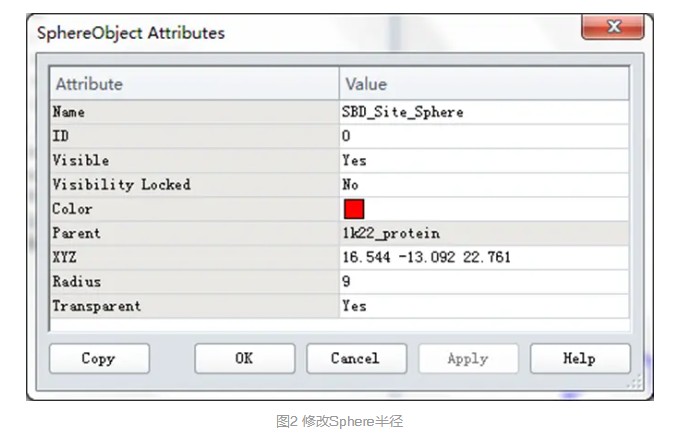
在视图窗口(Graphic View)中,选取红色的球体,或者系统视图(Hierarchy View)中选择SBD_Site_Sphere,右击选取Attributes of SBD_Site_Sphere。
打开SphereObjectAttributes对话框,将Radius设为9。(图2)
球体半径值可以调大以包含活性位点处的所有残基,但是这同样也会增加所要识别的相互作用的数量,其中可能包含一些非相关信息。
3. 产生相互作用模型
在受体活性位点球体中产生相互作用模型。
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Edit and Cluster Pharmacophore Features,在工具面板中单击Interaction Generation,打开Interaction Generation对话框。
设置Input Receptor为1k22_protein_ligands:1k22_protein。
设置Input Site Sphere为已定义的半径为9埃的球体坐标。
其余参数使用默认值。(图3)
点击Run运行作业。
等待作业完成。

作业完成以后,在视图窗口(Graphic View)中会自动插入产生的药效团特征。(图4)

基于结构药效团模型的编辑
初次构建相互作用模型时,20个药效团特征元素是一个比较合理的数目。
1. 隐藏所有特征元素
在工具浏览器(Tools Explorer)中,展开Pharmacophore | Edit and Cluster Pharmacophore Features,在工具面板中单击Show/Hide Location Spheres,隐藏feature周围的球。
注意矢量化的特征元素可以表征同某一个特定的氨基酸残基间的相互作用。
在系统视图(Hierarchy View)中,关闭SBD_Site_Sphere。
此时,视图窗口(Graphic View)中只有不同颜色标记的结合口袋,配体分子,分子间相互作用。
在Edit and Cluster Pharmacophore Features工具面板中,定义Current Feature为All Features。
单击Show/Hide All Features,隐藏所有特征元素。
2. 对feature进行聚类
首先定义Current Feature为Acceptor,点击Edit and Cluster Pharmacophore Features工具面板中的Show/Hide Acceptor,显示所有氢键受体特征。
点击Edit and Cluster Pharmacophore Features工具面板中的Cluster Current Features,对Acceptor特征进行聚类分析。
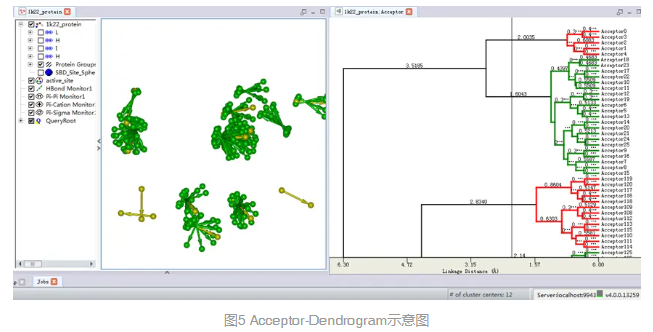
自动出现一个新的窗口,包含聚类分析的树形图。(图5)
将该Dendrogram窗口标签拖至视图窗口使得该窗口同1k22_protein_ligand分子窗口并列显示。
注:在该窗口中滚动鼠标滚轮,可以纵向调节图形大小,方便观察所有树形分支。移动该窗口中的slider线,显示不同的聚类中心。相应的该聚类中心会在分子窗口中高亮显示。该聚类是基于所有特征元素间的几何距离来定义的。自动聚类分析和手工选取相结合可以方便用户选取更佳特征元素的组合来表征蛋白活性位点。
移动窗口中的slider线,直到窗口右下角显示Number of cluster centers为12。
在工具面板中点击Keep Only Cluster Centers,关闭Dendrogram Window。
这样所有的Acceptor feature将由12个Acceptor feature代表。
在系统视图(Hierarchy View)中,展开QueryRoot | Fit Fit_1。
CTRL-选取acceptor feature28,46,56,77,97,单击工具面板中的Keep Only Current Feature Selections,以保留这些Acceptor特征。
选取这些特定元素特征是因为这几个Acceptor特征对应了活性位点的特定氨基酸残基,而其他特征则要么离残基比较远要么取向偏离了可能的结合位点。
单击工具面板中的Show/Hide Acceptor,隐藏Acceptor特征。
设置工具面板中的Current Feature为Donor,点击Show/Hide Donor,显示所有氢键供体特征。
在系统视图(Hierarchy View)中,展开QueryRoot | Fit Fit_1。
CTRL-选取donor feature44,63,75,76,94,101,107,141,198,207,单击工具面板中的Keep Only Current Feature Selections,以保留这些Donor特征。
手动选取这些特定元素特征作为cluster center的代表同活性位点重要残基相互作用。这些特征的选取仅仅是依据基于蛋白结构信息得到的相互作用模型,用户也可以依据已知的氢键相互作用来手工选取。
单击工具面板中的Show/Hide Donor,隐藏Donor特征。
设置工具面板中的Current Feature为Hydrophobe,点击Show/Hide Hydrophobe,显示所有疏水特征。
在系统视图(Hierarchy View)中,展开QueryRoot | Fit Fit_1。
CTRL-选取hydrophobe feature9,48,81,单击工具面板中的Keep Only Current Feature Selections,以保留这些Hydrophobe特征。
设置工具面板中的Current Feature为All Features,点击Show/Hide All Features,显示所有保留特征元素。
蛋白活性位点处药效团模型共由18个特征元素表征,特征元素的选取是基于观察到的配体和蛋白间相互作用,以及从单一蛋白结构得到的作用。
注:另一个用于编辑药效团模型的重要工具是Average Pharmacophore Features命令。选取同一类型的特征元素,再用该命令基于这些特征元素得到一个平均特征用以代表整个类别。
在系统视图(Hierarchy View)中,CTRL-选取acceptor feature28,77,97,donor feature63,75,141,198,从菜单栏中选取Edit | Group。
命名为S-interactions。
该步骤所定义的相互作用都表征的是S-口袋处的残基。
3. 产生排除体积模型(exclusion model)
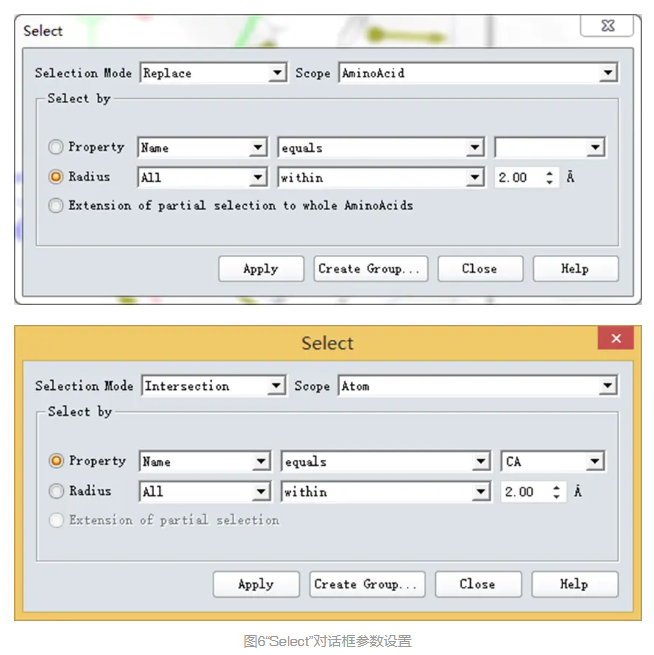
在系统视图(Hierarchy View)中,选择QueryRoot,然后在主菜单栏中选择Edit | Select。在Select对话框中,点击Selection Mode右边的栅格,下拉列表中选择Replace,点击Scope右边的栅格,下拉列表中选择AminoAcid。
选取Radius,设为2.00,点击Apply按钮。
点击Selection Mode右边的栅格,下拉列表中选择Intersection,点击Scope右边的栅格,下拉列表中选择Atom。
选取Property,在最后一栏下拉菜单中选取CA,点击Apply。(图6)
保留的特征元素周围2埃以内完整残基中的CA原子被选中。
从主菜单栏中选取Structure | Query Features,将Feature改设为ExclusionSphere,基于选取的原子产生排除体积。(图7)

通过以上步骤,最终得到了一个基于蛋白结构的药效团模型,含有排除体积。(图8)

配体库的筛选
在用该药效团模型筛选化合物库之前有必要对该模型进行验证,考察这18个药效团特征元素中那些特征元素比较关键。
本教程通过对一个包含了活性分子和非活性分子的测试集进行虚拟筛选来对该模型进行验证。
1. 采用药效团进行数据库筛选
在文件浏览器(File Explorer)中,展开Samples | Tutorials | Pharmacophore,双击打开thrombin_ligands.sd。
读入8个有活性的凝血酶抑制剂,分子名都有_ligand的后缀。
接着展开Samples | Tutorials | Receptor-Ligand Interaction,将kinase_ligands.sd文件拖至thrombin_ligands窗口中。
读入9个激酶抑制剂,这些分子都视为未知配体(理想情况下,这些分子应确证不能同凝血酶结合才可视为负性对照物)。
在系统视图(Hierarchy View)中,选中Hydrophobe81特征作为筛选过程中必须出现的特征元素。
在工具浏览器(Tool Explorer)中,展开Pharmacophores | Search, Screen an Profile,点击Screen Library,打开Screen Library对话框。
设置Input Ligands为thrombin_ligands:All。
确保Input Pharmacophore为1k22_protein_ligand: QueryRoot。
接着指定筛选得到的配体分子必需匹配的特征元素个数,以及用于筛选的每个药效团模型中都必须包含的特征元素。
在Screen Library对话框中,展开Input Pharmacophore参数组。
点击Required Features右边的栅格,下拉列表中选择True,并展开该参数组,设置Required Features为前面选中的Hydrophobe81。

点击Features Group右边的栅格,下拉列表中选择True,并展开该参数组,设置Feature Group 1为前面定义的S-interactions。
展开Feature Group 1参数组,将Maximum改为7。
该值为该特征元素组中特征元素的上限个数。
将Maximum Features设为8。
该值为输入药效团模型中需要考虑的特征元素的上限个数。
展开Input Ligands参数组,点击Conformation Generation右边的栅格,下拉列表中选择FAST。
展开Advanced参数组,查看Number of Possible Pharmacophores。
该值给出了药效团模型几种组合的可能性,是基于Total Number of Features,Minimum Features和Maximum Features这三个参数计算得来的,该值当前显示为105774。
以上参数的设置表明最终筛选得到的配体小分子必须同编辑得到的18个特征元素中的4个特征元素相匹配,并且在这4个特征元素当中必须有一个是Hydrophobe81特征,同时还必须有一个特征来自于特征元素组S-interactions。
要想正确运行该流程还必须设置Maximum Subset of Pharmacophores参数以确保能够搜索到蛋白活性位点的所有药效团空间。
将Maximum Subset of Pharmacophores改为250。
其余参数设诶默认设置。(图9)
点击Run运行作业。
等待作业完成。
2. 分析查看筛选结果
作业完成后,自动打开Report页面,点击View All Results链接,打开一个新的分子窗口。
该窗口中包含了一个structure-based pharmacophore和同所有类别的药效团匹配更好的命中分子。一共筛选得到55个分子,包含了17个凝血酶和激酶抑制剂中的11个配体分子。
在表格浏览器(Data Table)中,单击右键,选取Filter …,打开Specify Filter对话框。
将FitValue的Choice选取为Top(#),Value选取为1。
勾选Apply filter to each group of identical value的复选框,选择Name。(图10)
针对表格中Name相同的分子只保留FitValue最高的一个。
可以看到8个凝血酶抑制剂中筛出了7个,9个激酶抑制剂中筛出了4个。

在表格浏览器(Data Table)中,单击右键,选取Remove Filters,还原前步的设置。
双击Name一栏使得凝血酶抑制剂排在表格的前面,不选择表中任何一栏。
从主菜单栏中选取Chart | Heat Map,跳出Choose Plot Axes对话框,设定热图的X轴和Y轴属性。
针对Y轴,选取Name属性。
针对X轴,选取Acceptor28,接着SHIFT-选取Hydrophobe9。
将ConfNumber和FitValue属性的选取取消。(图11)
即纵坐标为Name,横坐标为所有共18个药效团特征元素。

坐标设定完以后,自动生成热图。(图12)
从该图可以观察到18个药效团特征元素分别同每个分子的匹配情况。例如,Hrdrophobe81该特征同所有分子都相互匹配,正如前面所设定的。而Donor94则只同很少的分子相匹配,因此当筛选更大的分子库时可以考虑删除该特征。





 苏公网安备 32059002002276号
苏公网安备 32059002002276号